Болезнь Гоше
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
В статье представлены данные по эпидемиологии, патогенезу, современной классификации и основным клиническим проявлениям болезни Гоше у детей. Приведены критерии дифференциальной диагностики с другими заболеваниями. Показано, что единственным эффективным методом лечения болезни Гоше служит патогенетическая ферментозаместительная терапия, которая купирует основные клинические проявления болезни, улучшая качество жизни больных и не оказывая выраженных побочных эффектов. Для такого лечения используют имиглюцеразу, под действием которой происходит гидролиз гликолипида глюкоцереброзида до глюкозы и церамида по обычному пути метаболизма мембранных липидов. Имиглюцераза показана для длительной заместительной ферментотерапии у больных с подтвержденной болезнью Гоше 1-го и 3-го типа. Рекомендовано мониторирование состояния больных на фоне терапии в соответствии с требованиями Объединенной международной группы по изучению болезни Гоше. Указаны основные ошибки диагностики и ведения таких пациентов, а также необоснованные назначения при лечении этого заболевания.
Ключевые слова
Об авторах
Научный центр здоровья детей РАМН, Москва
Россия
кандидат медицинских наук, заведующая отделением восстановительного лечения детей с болезнями органов
пищеварительной системы НИИ профилактической педиатрии и восстановительного лечения ФГБУ «Научный центр здоровья детей» РАМН
Научный центр здоровья детей РАМН, Москва, Российская Федерация Первый Московский государственный медицинский университет им. И.М. Сеченова, Российская Федерация Российский национальный исследовательский медицинский университет имени Н.И. Пирогова, Москва, Российская Федерация
Россия
Научный центр здоровья детей РАМН, Москва, Российская Федерация Первый Московский государственный медицинский университет им. И.М. Сеченова, Российская Федерация
Россия
Первый Московский государственный медицинский университет им. И.М. Сеченова, Российская Федерация
Россия
Список литературы
1. Mankin H. J., Rosenthal D. I., Xavier R. Gaucher disease. New approaches to an ancient disease. J. Bone Joint Surg. Am. 2001; 83A: 748–762.
2. Koprivica V. V., Stone D. L., Park J. K., Callahan M., Frisch A., Cohen I. J. et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000; 66 (6): 1777–1786.
3. Pastores G. M., Weinreb N. J., Aerts H., Andria G., Cox T.M., Giralt M. et al. Therapeutic goals in the treatment of Gaucher disease. Supp. Semin. Hematol. 2004; 41 (5): 4–14.
4. Andersson H., Kaplan P., Kacena K., Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008; 122 (6): 1182–1190.
5. Weinreb N. J., Taylor J., Cox T., Yee J., vom Dahl S. A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am. J. Hematol. 2008; 83: 890–895.
6. Атлас редких болезней. под ред. А.А. Баранова, Л.С. НамазовойБарановой. М.: ПедиатрЪ. 2013. 304 с.
7. Wenstrup R. J., Kacena K. A., Kaplan P., Pastores G. M., Prakash Cheng A., Zimran A. et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type I Gaucher disease. JBMR Reprint. 2007; 22 (1): 119–126.
8. Wenstrup R. J., Roca-Espiau M., Weinreb N. J., Bembi B. Skeletal aspects of Gaucher disease: a review. Br. J. Radiol. 2002; 75 (1): 2–12.
9. Bembi В., Ciana G., Mengel E., Terk M. R., Martini C., Wenstrup R. J. Bone complications in children with Gaucher disease. Br. J. Radiol. 2002; 75 (1): 37–43.
10. Гундобина О. С., Малахов О. О., Морев С. Ю., Малахов О. А. Развитие костной патологии при болезни Гоше 1-го типа у детей. Клинический пример. Доктор Ру. 2011; 5 (64): 10–14.
11. Vellodi A., Bembi B., de Villemeur T. B., Collin-Histed T., Erikson A., Mengel E. et al. Management of neuronopathic Gaucher disease: а European consensus. J. Inherit. Metab. Dis. 2001; 24 (3): 319–327.
12. Baldellou A., Andria G., Campbell P. E., Charrow J., Cohen I. J., Grabowski G. A. et al. Paediatric non-neuronopathic Gaucher disease: recommendations for treatment and monitoring. Eur. J. Pediatr. 2004; 163 (2): 67–75.
13. Темин П. А., Казанцева Л. З. Наследственные нарушения нервно-психического развития детей. Руководство для врачей. М.: Медицина. 2001. 432 с.
14. Краснопольская К. Д. Наследственные болезни обмена веществ. Справочное пособие для врачей. М.: Центр социальной адаптации и реабилитации детей «Фохат». 2005. 364 с.
15. Grabowski G. A., Hopkin R. J. Enzyme therapy for lysosomal storage disease: Principles, practice, and prospects. Annu. Rev. Genomics. Hum. Genet. 2003; 4: 403–436.
16. Weinreb N. J., Goldblatt J., Villalobos J., Charrow J., Cole J. A., Kerstenetzky M., vom Dahl S., Hollak C. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab. Dis. 2012 Sep. 14 [Epub ahead of print].
Болезнь Гоше: что это
Болезнь Гоше иначе называют сфинголипидозом или глюкозилцерамидным липидозом. Это заболевание носит наследственный характер и вызвано генетической патологией. Как отдельное заболевание болезнь Гоше была открыта в 1882 году французским врачом Филиппом Гоше. Ему довелось лечить пациентку, у которой были выявлены специфические изменения в тканях селезенки. Спасти женщину не удалось в связи с заражением крови.
Через несколько десятилетий господин Гоше столкнулся с аналогичным клиническим случаем. Специалисту удалось определить, что в тканях селезенки накапливается глюкоцереброзид и этот процесс сопровождается недостаточностью фермента глюкоцереброзидазы. На основании результатов обследования специалист предположил, что данные изменения связаны с генетической патологией.
Как явление болезнь Гоше встречается достаточно редко. Из 70 тысяч испытуемых мутационный ген был обнаружен лишь у одного человека. При этом замечено, что у некоторых народов распространенность такого заболевания в несколько десятков раз выше. Это обусловлено историко-культурными традициями, которые допускают браки между людьми, состоящими в родстве.
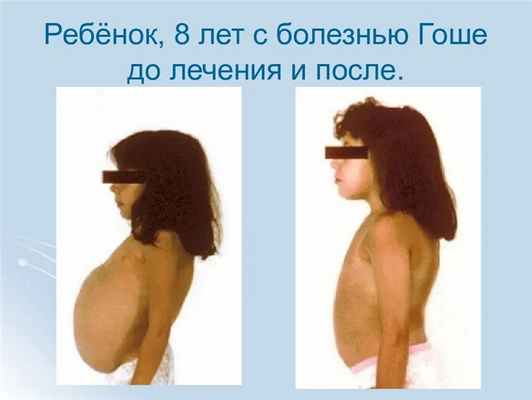
Особенности заболевания и риск передачи потомству
Болезнь Гоше относится к разряду наследственных ферментопатологий. Из-за недостаточной выработки лизосомальных ферментов нарушается жировой обмен. Лизосомы представляют собой клеточные органеллы – компоненты клеток, которые содержат несколько десятков гидролитических ферментов. Они необходимы для расщепления белков, жиров, углеводов, нуклеиновых кислот. Из-за неправильной работы лизосом возникают так называемые болезни накопления, при которых в клетках накапливаются липиды, мукополисахариды, гликопротеины. Наследственных болезней накопления на сегодняшний день известно более пятидесяти.
При болезни Гоше из – за недостаточности фермента бета-глюкозидазы нарушается расщепление гликосфинголипидов – соединения липидов и углеводов. В норме фермент отщепляет глюкозу от глюкоцереброзида. Если этого не происходит, макромолекулы накапливаются в клеточных структурах, образуя так называемые клетки Гоше. Их иначе называют пенистыми клетками из-за характерной формы. При нарушении обмена жиров начинаются расстройства в работе различных органов и систем. Сильнее всего страдают печень, селезенка, костный мозг, опорно-двигательный аппарат.
Болезнь наследуется аутосомно – рецессивным способом. Если у обоих родителей присутствует дефектный ген, риск рождения ребенка с ферментной патологией составляет 25%. Вероятность того, что новорожденный будет носителем поврежденного гена, достигает 50%, если мутация есть только у одного родителя или у обоих.
Парам, чьи близкие родственники больны болезнью Гоше, показано медико-генетическое консультирование. В первом триместре беременности проводят анализ амниотической жидкости, чтобы определить уровень активности фермента у плода. При обнаружении патологии женщине предлагается прервать беременность.
Симптомы болезни Гоше
Симптомы заболевания определяются типом болезни Гоше. Существует три разновидности патологии. Общими для всех типов являются увеличение в размерах печени и селезенки, что мешает их нормальной работе, нарушения в системе кроветворения. В остальном симптоматика и возраст проявления патологии зависят от вида заболевания.
Первый тип
Эта разновидность является самой распространенной. Сразу после рождения у ребенка начинается постепенное увеличение печени и селезнки, однако клинические проявления патологии наступают позже. У некоторых пациентов болезнь проявляется только к 30 – 40 годам.
Нарушения со стороны системы кроветворения проявляются анемией, снижением уровня тромбоцитов, кровоточивостью. Пациенты жалуются на боли в костях и суставах. У детей могут наблюдаться деформационные изменения, из – за которых нарушается работа опорно-двигательного аппарата. Также болезнь негативно влияет на рост ребенка: обычно ребенок с заболеванием Гоше вырастает ниже среднего роста. Из-за негативного влияния заболевания на костную ткань пациенты с болезнью Гоше подвержены остеопорозу, переломам.
У взрослых пациентов заболевание может проявляться повышенной пигментацией кожи. На лице и ногах появляются желтые и коричневые пятна, в области вокруг глаз могут возникать покраснения.
Второй тип
Он встречается редко и характеризуется проявлением в раннем детском возрасте. Как правило, это происходит в первые полтора года жизни.
Как и при первом типе, ферментная патология проявляется увеличением печени и селезенки. У новорожденного нарушены глотательные рефлексы, могут появиться проблемы с дыханием. Клетки Гоше формируются в ЦНС, что проявляется неврологическими расстройствами.
Ребенок заметно отстает в физическом и психическом развитии. До года у него снижен тонус мышц, а затем возникает гипертонус. Он сильнее всего проявляется в области шеи и конечностей, ограничивает подвижность, вызывает боли. Гипертонус приводит к спазмам, параличу. Из неврологических проявлений можно отметить судороги. У многих пациентов развивается косоглазие. Дети с болезнью Гоше второго типа подвержены пневмонии, которая протекает в тяжелой форме.
Особенностью патологии является то, что она плохо поддается лечению и приводит к тяжелой инвалидности с детства, ранней гибели пациента.
Третий тип
Первые симптомы болезни проявляются в 2 – 3 года. При обследовании обнаруживают увеличение печени и селезенки. В детском или подростковом возрасте начинается поражение ЦНС, что вызывает характерные изменения. У ребенка возникают спазмы мышц и судороги, развивается косоглазие, появляется гипертонус мышц, могут наблюдаться проблемы с глотанием и дыханием.
Постепенно снижаются познавательные функции, ребенок теряет способность говорить, писать. Болезнь проявляется эмоциональной неустойчивостью, вплоть до психозов. Подростки отстают в половом развитии.
Особенность болезни Гоше третьего типа заключается в быстром прогрессировании и ухудшении состояния пациента.
Осложнения сфинголипидоза
Без лечения пациенты могут погибнуть из-за серьезных нарушений в работе печени и селезенки, в которых в первую очередь накапливаются клетки Гоше. Наиболее опасными для жизни являются второй и третий тип заболевания, при которых поражается ЦНС. У пациентов происходят изменения в структуре спинного и головного мозга. Спазм гортани и нарушения дыхательной функции могут привести к острой кислородной недостаточности и смерти от удушья. Из-за снижения уровня тромбоцитов возрастает риск кровотечения, которое из-за большого масштаба опасно для жизни.
При первом типе сфинголипидоза в первую очередь страдает опорно-двигательная система. У пациента разрушается костная ткань, теряется ее плотность. В случае травмы это приводит к серьезным переломам. Также могут наблюдаться инфекционные поражения костей .
Из-за спазмов и паралича пациент испытывает сложности и самостоятельным передвижением, нуждается в постоянной помощи и уходе.
Диагностика
При подозрении на наследственную ферментную патологию необходимо пройти обследование у целого ряда специалистов: генетика, эндокринолога, гематолога, психиатра, офтальмолога, невролога. Следует понимать, что по некоторым симптомам болезнь Гоше совпадает с костным туберкулезом, вирусным гепатитом, гемобластозом и другими заболеваниями, поэтому для постановки диагноза назначают обширный комплекс процедур.
Общеклинический и биохимический анализ крови
По результатам лабораторного исследования обнаруживается снижение гемоглобина, лейкоцитов и тромбоцитов. По результатам биохимии можно определить, что активность глюкоцереброзидазы понижена.
Ферментный анализ клеток
В качестве материала для исследования используются образцы сухой крови и фибробласты кожи. При болезни Гоше обнаруживают сниженную активность фермента глюкозидазы при ярко выраженном повышении активности фермента под названием хитотриозидаза.
Исследование костной ткани и костного мозга
Чтобы исключить онкологические заболевания системы кроветворения, пациенту проводят исследование костного мозга. При болезни Гоше в нем обнаруживают характерные для этой патологии клеточные структуры.
Для оценки состояния костной ткани назначают денситометрию, МРТ, рентген. У пациента наблюдаются остеопения и остеопороз, очаги остеонекроза.
Диагностика внутренних органов
Самый первый маркер болезни Гоше – увеличение печени и селезенки. Для обнаружения патологии проводят УЗИ органов брюшной полости или МРТ.
Генетический анализ
Если по результатам комплексной диагностики не удается точно определить наличие патологии, назначается специальный ДНК – тест, который подтвердит или опровергнет наличие дефектного гена.
Лечение болезни Гоше
Терапия болезни Гоше в первую очередь направлена на восполнение ферментной недостаточности. Пациент постоянно принимает препараты на основе рекомбинантной глюкоцереброзидазы, которая вводится при помощи внутривенных инъекций. Такое лечение дает положительный эффект при первом и третьем типах заболевания. У некоторых пациентов развитие болезни удается остановить, они могут вести обычный образ жизни.
В США и европейских странах людям с умеренными проявлениями заболевания назначают субстрат – редуцирующую терапию. В качестве лекарств используют ингибиторы глюкозилцерамидсинтазы. Их применение уменьшает образование субстрата гликосфинголипидов и замедляет их накопление за счет ускорения катаболизма макромолекул.
Лечение болезни Гоше также включает лекарственную терапию, которая замедляет разрушение костной ткани. В первую очередь это препараты кальция и витамина D, продукты, богатые этим минералом. При инфекционных поражениях костей показана антибактериальная терапия. Для снятия болей назначают нестероидные противовоспалительные препараты.
Для уменьшения неврологической симптоматики используются миорелаксанты, ноотропные препараты.
При втором типе болезни Гоше необходимо следить за дыхательной активностью, своевременно устранять сбои, которые могут приводить к остановке дыхания.
Некоторым пациентам со сфинголипидозом проводится хирургическое удаление селезёнки, пересадка костного мозга.
Продолжительность жизни при болезни Гоше
Наиболее благоприятный исход характерен для первого типа заболевания. Он проявляется поздно и лучше других поддается лечению, что позволяет пациенту дожить до зрелого возраста. При третьем типе продолжительность жизни большинства пациентов ранее составляла 15 – 17 лет, но благодаря усовершенствованию лечения многим удается доживать до 30 – 40 лет.
Самый неблагоприятный прогноз характерен для второго типа. Патологические изменения появляются в первые месяцы жизни, и болезнь быстро прогрессирует. Большинство заболевших умирает в течение первых двух лет жизни.
Болезнь Гоше
Болезнь Гоше является сфинголипидозом Сфинголипидоз Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты клетки), либо захваченные извне. Унаследованные дефекты или недостатки. Прочитайте дополнительные сведения – наследственным нарушением метаболизма, которое возникает в результате дефицита глюкоцереброзидазы и приводит к накоплению глюкоцереброзида и родственных соединений. Симптомы и признаки варьируются в зависимости от типа, но чаще всего – гепатоспленомегалия или изменения центральной нервной системы. Диагноз ставят на основании анализа ДНК и/или анализа ферментов лейкоцитов. Лечение состоит в ферментозамещающей терапии глюкоцереброзидазой
Для получения дополнительной информации см. таблицу Некоторые сфинголипидозы Некоторые сфинголипидозы .
Глюкоцереброзидаза обычно гидролизует глюкоцереброзид до глюкозы и керамидов. Генетические дефекты фермента вызывают накопление глюкоцереброзида в тканевых макрофагах через фагоцитоз, образуя клетки Гоше. Накопление клеток Гоше в перивас-кулярных пространствах в мозге вызывает глиоз при нейропатических формах.
Выделяют 3 типа болезни Гоше, различающиеся по эпидемиологии, активности фермента и проявлениям.
Болезнь Гоше I типа
Тип I (ненейропатический) является наиболее распространенным (90% всех больных). Остаточная активность фермента максимальна. Евреи-ашкенази подвергаются наибольшему риску; 1/12 являются носителями. Возраст возникновения заболевания колеблется от детского до зрелого взрослого.
Симптомы и признаки болезни Гоше I типа включают спленогепатомегалию, костные заболевания (например, остеопению, болевые кризы, остеолитические повреждения с переломами), задержку роста, задержку полового созревания, кровоподтеки и пингвекулу. Носовые кровотечения и синяки в результате тромбоцитопении являются общими проявлениями.
Рентгенография показывает бросающиеся в глаза концы длинных костей (колбообразная деформация Эрленмейера) и кортикальное истончение.
Болезнь Гоше II типа
Тип II (острый нейропатический) является наиболее редким, и остаточная активность фермента при этом типе самая низкая. Начало заболевания приходится на период младенчества.
Симптомами и признаками болезни Гоше II типа являются прогрессирующим неврологическим ухудшением (например, ригидность, судороги) и смерть наступает в возрасте 2 лет.
Болезнь Гоше III типа
Тип III (подострый нейропатический) размещается между типами I и II по распространенности, активности ферментов и клинической тяжести. Заболевание возникает в любое время в детском возрасте.
Клинические проявления зависят от подтипа и включают прогрессирующее слабоумие и атаксию (IIIa), поражение костей и органов (IIIb) и надглазничный паралич с помутнениями роговицы (IIIc). Пациенты, которые доживают до подросткового возраста, могут жить в течение многих лет.
Диагностика болезни Гоше
Определение активности ферментов
Диагноз болезни Гоше устанавливается на основании анализа ДНК и/или ферментного анализа лейкоцитов. Выявление носителей и определение типов проводится при помощи анализа мутаций. Хотя биопсия не является необходимой, клетки Гоше – заполненные липидами макрофаги в печени, селезенке, лимфатических узлах или костном мозге, имеющие вид мятой тонкой бумаги – являются диагностическими. (Также см. Исследования при подозрении наследственных нарушений обмена веществ Начальное тестирование Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения ).
Лечение болезни Гоше
Типы I и III: замена фермента глюкоцереброзидазой
Иногда назначается миглустат, элиглустат, спленэктомия или трансплантация стволовых клеток или костного мозга
Замещение фермента внутривенным введением глюкоцереброзидазы эффективно при типах I и III, для типа II лечение отсутствует. Ферменты модифицируют для эффективной доставки в лизосомы. Пациенты, получавшие замещение фермента, требуют регулярного мониторинга уровня гемоглобина и тромбоцитов, регулярной оценки состояния селезенки и объема печени, КТ или МРТ, а также регулярной оценки состояния заболевания костей при исследовании скелета, двухэнергетической рентгеновской абсорбциометрии или МРТ.
Миглустат (100 мг перорально три раза в день), ингибитор глюкозилцерамид синтазы, снижает концентрацию глюкоцереброзида (субстрат для глюкоцереброзидазы) и является альтернативой для пациентов, которые не могут получить замещение фермента.
Элиглустат (84 мг перорально раз или два в день), другой ингибитор глюкозилцерамид синтазы также снижает концентрацию глюкоцереброзида.
Спленэктомия может быть полезна пациентам с анемией, лейкопенией, тромбоцитопенией, или когда размер селезенки вызывает дискомфорт. Пациентам с анемией может потребоваться переливание крови.
Трансплантация костного мозга или стволовых клеток обеспечивает окончательное излечение, но рассматривается только в крайних случаях из-за значительной заболеваемости и смертности.
Ключевые моменты
Болезнь Гоше является сфинголипидозом, развивающимся в результате дефицита глюкоцереброзидазы, который приводит к накоплению глюкоцереброзида.
Выделяют 3 типа, различающиеся по эпидемиологии, активности фермента и проявлениям.
Симптомы и признаки варьируются в зависимости от типа, но чаще всего – гепатоспленомегалия или изменения центральной нервной системы.
Диагноз болезни Гоше устанавливается на основании анализа ДНК и/или ферментного анализа лейкоцитов. Носителей выявляют и типы различают при помощи анализа мутаций.
Лечение для типов I и III, включают в себя замещение фермента глюкоцереброзидазой, а иногда и назначение миглустата, элиглустата, спленэктомии или трансплантации стволовых клеток или костного мозга; нет никакого лечения для типа II.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Online Mendelian Inheritance in Man® (OMIM®) database: полная информация о генах и их молекулярной и хромосомной локализации
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Болезнь Гоше
Образование: 2009 г. – Сибирский федеральный университет, специальность «Психология» 2012 г. – Красноярский государственный медицинский университет имени профессора В.Ф. Войно-Ясенецкого, специализация по клинической психологии. Професиональные навыки: Специализируется на дифференциальной диагностике психических расстройств, коррекционной работе с детьми с расстройствами аутистического спектра. Владеет навыками проведения психодиагностики, психологического консультирования, психокоррекции. О себе: В своей работе придерживаюсь принципов открытого диалога и разделения ответственности. Считаю, что достижение положительного результата в диагностике, консультировании или терапии возможно только при сотрудничестве врача и пациента.
Описание
Болезнь Гоше – генетическая патология, в основе которой лежит снижение функциональности фермента β-глюкоцереброзидазы, обеспечивающей распад продуктов метаболизма в клетке. Заболевание является наследственной ферментопатией, лизосомной болезнью накопления. Код в международной клаасификации болезней 10 пересмотра – Е 75.2.
Название болезнь Гоше получила по фамилии врача, описавшего ее впервые. Он определил тип клеток, которые патогномоничны для данной ферментопатии – макрофаги с накоплениями липидов. Позже в медицинском сообществе их стали называть клетками Гоше.
Болезнь считается редкой, встречается у 1 человека из 50 тысяч населения. Исключение составляет популяция евреев-ашкенази, среди них распространенность патологии – 1:450.
Наследование болезни Гоше – аутосомно-рецессивное. Основой являются мутации гена, который кодирует структуру молекулы фермента бета-глюкоцереброзидазы. Его локализация – регион q21, первая хромосома. Если обе аллели мутантные, то способность фермента расщеплять продукты обмена снижается на 30%. В итоге лизосомы макрофагов накапливают молекулы липидов. Это приводит к хронической активации макрофогальной системы: печень, селезенка и костный мозг становятся увеличенными из-за скопления патологических клеток. У больных развивается спленомегалия (увеличение селезенки), гепатомегалия (увеличение печени), инфильтрация костного мозга, поражение костно-суставной системы.
Симптомы
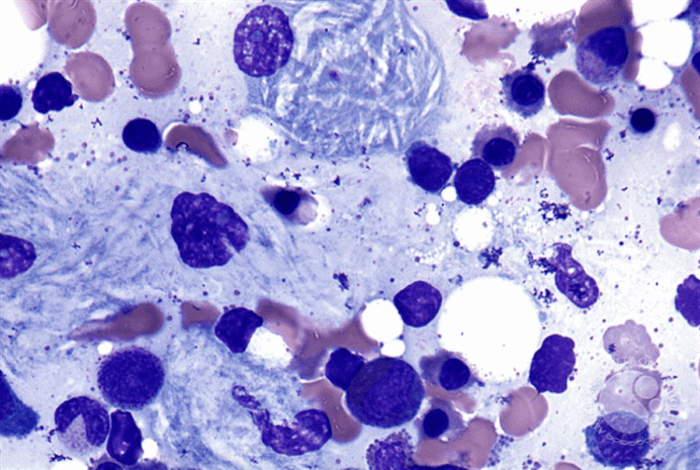
Выделено три варианта течения заболевания:
- Без неврологических симптомов.
- С быстро прогрессирующими неврологическими симптомами.
- С хроническим усилением неврологических симптомов.
Болезнь Гоше первого типа – самый распространенный вариант (90% больных). Наблюдается прогрессирующее увеличение селезенки и печени – органов, основу которых составляет паренхиматозная ткань. Развивается нарушение костной системы и панцитопения – дефицит всех клеток крови: эритроцитов, лейкоцитов и тромбоцитов. Болезнь может проявить себя в любом возрасте, распространенность среди мужчин и женщин одинаковая.
Дети отстают в физическом и половом развитии: они ниже ростом, менее ловки и быстры, чем сверстники, позже переживают пубертат. Наиболее ранний признак болезни Гоше 1 типа – увеличение селезенки (спленомегалия). Орган становится плотным, прощупывается пальцами в области левого подреберья. Увеличение печени менее выраженное. Этот орган прощупывается на более поздних стадиях заболевания.
Поражение костей скелета – причина инвалидизации больных. Структура костной ткани нарушается из-за замещения нормальных клеток костного мозга клетками Гоше. Первыми поражаются кости бедер, затем – другие трубчатые кости, в последнюю очередь – позвоночник. У многих больных масса тела снижается вследствие уменьшения минеральной плотности костей. Развивается отек, увеличивается внутрикостное давление, появляются острые костные боли (костные кризы).
В результате быстрого проникновения клеток Гоше в костный мозг происходит сосуды сужаются, их стенки склеиваются, образуются тромбы. В костной ткани развиваются очаги остеонекроза. Изменяется строение головок бедренных костей, нарушаются функции нижних конечностей. Боли в костях усиливаются. Больным может потребоваться операция (эндопротезирование сустава).
Болезнь Гоще второго типа имеет самое тяжелое течение. Она проявляется в первый год жизни. У детей увеличивается в размерах печень и селезенка, то есть развивается гепатоспленомегалия. Наблюдаются окуломоторные аномалии: непроизвольные колебательные движения глазных яблок (нистагм) и частичная их неподвижность (офтальмопарез). Нередко отмечается непроизвольное сокращение и спазм мускулатуры челюстей (тризм), двустороннее косоглазие, спастичность мышц.
После 6 месяцев прогрессирует задержка в психомоторном развитии: ребенок позже начинает сидеть, слабо лепечет, речь отсутствует либо представлена единичными словами. На поздних стадиях распадаются ранее приобретенные навыки. Часто нарушается глотание, дети вдыхают еду, возникает аспирационная пневмония. Развиваются тонико-клонические судорожные приступы, которые не купируются противосудорожными препаратами. Заболевание быстро прогрессирует и завершается летальным исходом на 1-2 году жизни.
Болезнь Гоше третьего типа проявляется в раннем детстве либо после 10 лет. Характерен замедленный характер прогрессирования. На начальных этапах увеличиваются в размерах печень и селезенка, проявляются неврологические нарушения аналогичные таковым при втором типе болезни. Основной признак патологии – миоклонические судороги, проявляющиеся как быстрые подергивания нескольких групп мышц. Они развиваются ка при нагрузке, так и в покое, с течением заболевания становятся более частыми и навязчивыми. На поздних стадиях миоклонические судороги сменяются генерализованными клоническими судорогами (патологическим напряжением всех мышц тела).
Интеллектуальные расстройства при болезни Гоше 3 типа варьируют от легкого снижения до грубой деменции с распадом всех навыков. Мозжечковые симптомы проявляются нарушением равновесия, способности удерживать позу. Возможно нарушение речи, письма, социального поведения. Иногда наблюдаются психотические эпизоды. Из-за прогрессирующих неврологических симптомов болезнь приводит к смерти. Средняя продолжительность жизни больных – 13-17 лет, однако известны случаи, когда пациенты проживали до 30-40 лет.
Диагностика
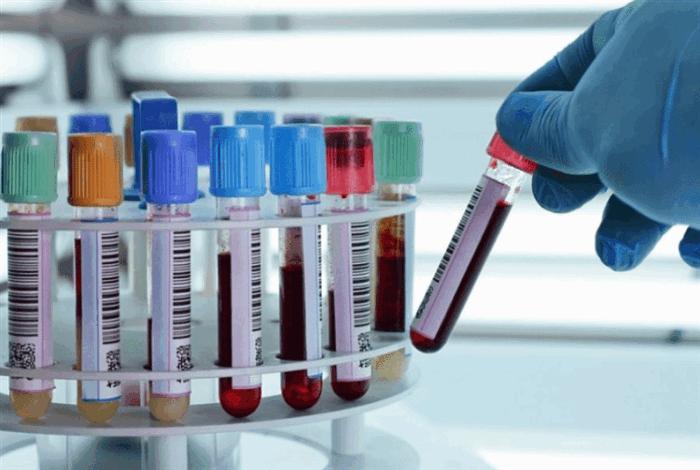
Подозрение на болезнь Гоше возникает у врачей при выявлении сплено- и гепатомегалии, не объяснимых более распространенными причинами, чем генетическое заболевание. Основой для постановки диагноза являются результаты лабораторных тестов, но всем больным показано комплексное обследование, которое включает:
- Сбор анамнеза и физикальные методы. При первичной беседе врач может выявить:
- задержку физического, полового и психического развития;
- увеличенные размеры селезенки и печени при пальпации;
- жалобы слабость, утомляемость, частые респираторные инфекции;
- признаки геморрагического синдрома (кровоточивость слизистых оболочек, подкожные гематомы);
- жалобы на боли в костях и суставах, переломы костей;
- косоглазие, апраксию, гиперрефлексию, спастичность и другие неврологические нарушения;
- интеллектуальное снижение, утрату навыков;
- наличие болезни Гоше и ее симптомов у близких родственников.
- Лабораторные исследования. Основной лабораторный признак заболевания – сниженная ферментативная функция β-глюкоцереброзидазы (β-GCase) в лейкоцитах. В качестве биоматериала может выступать не только цельная кровь, но и ее высушенные пятна, культура клеток кожных фибробластов. Еще один лабораторный признак – повышенная концентрация хитотриозидазы (гидролитического фермента, синтезируемого макрофагами). У 94% пациентов показатель выше нормы в 10-30 раз. Исследование ДНК не является обязательным, но иногда необходимо. При морфологическом анализе костного мозга обнаруживаются клетки Гоше.
- Инструментальные процедуры. Чтобы определить, насколько сильно повреждены костно-суставная система выполняется рентгенография. На снимках определяется изменение костной ткани: признаки распространенного остеопороза, изменение формы большеберцовых костей, локальное отмирание ткани. МРТ костей скелета и денситометрия на ранних этапах болезни выявляют изменение структуры костного мозга и остеопению. МРТ и УЗИ селезенки и печени необходимы для определения объемов органов, обнаружения очаговых поражений.
Лечение

Используется ферментозаместительная терапия. Это единственный эффективный метод лечения, благодаря которому купируются основные симптомы и улучшается жизнь больных. При этом нет тяжелых побочных реакций.
Цели ферментозаместительной терапии:
- устранение или уменьшение выраженности цитопении (дефицита клеток крови);
- купирование суставных и костных болей;
- нормализация размера печени и селезенки;
- предупреждение необратимого повреждения костей, суставов и внутренних органов.
Основой препаратов для ферментозмаестительной терапии является имиглюцераза. Ее производят методом ДНК-рекомбинантной технологии. Под воздействием имиглюцеразы гликолипида глюкоцереброзида гидролизируется до конечных продуктов распада – церамида и глюкозы. Биохимические процессы в лизосомах происходят по стандартному пути как у здоровых людей.
Препараты на основе имиглюцеразы показаны в качестве продолжительного лечения пациентам, у которых подтвержден диагноз «болезнь Гоше» типа I либо типа III (без вовлечения ЦНС либо с ее хроническим поражением). Все препараты вводятся внутривенно капельно.
Согласно клиническим исследованиям, у больных первым типом болезни Гоше, получающих имиглюцеразу на протяжении 10 лет, наблюдается значительное и стойкое улучшение состояния, что подтверждается восстановлением нормальной концентрации тромбоцитов и белка гемоглобина, нормализацией размеров печени и селезенки (по данным УЗИ), а также исчезновением костных и суставных болей.
При болезни Гоше второго типа использование ферментозаместительной терапии неэффективно, так как имиглюцераза не способна проникнуть через гематоэнцефалический барьер, то есть не поступает из крови в нервную ткань. Больным показана симптоматическое лечение. Все еще не существует методов для эффективной помощи детям в болезнью Гоше типа II.
Лекарства
Симптоматическая лекарственная терапия болезни Гоше нацелена на уменьшение проявлений остеопороза. Больным назначаются бисфосфонаты, альфакальцидол, соли кальция. Благодаря этим препаратам замедляется или полностью прекращается потеря костной массы, повышается прочность костей, предотвращаются переломы. Для уменьшения болей используются анальгетики.
Народные средства
Болезнь Гоше требует высококвалифицированной медицинской помощи и применения современных лекарственных препаратов для ферментозаместительного лечения. Народные методы при данном заболевании неэффективны, так как патологические процессы обусловлены генетической мутацией и дефицитом фермента.
Информация носит справочный характер и не является руководством к действию. Не занимайтесь самолечением. При первых симптомах заболевания обратитесь к врачу.
Болезнь Гоше
БГ — болезнь Гоше
ген GBA — ген глюкоцереброзидазы
ЗФТ — заместительная ферментная терапия
МРТ — магнитно-резонансная томография
ЦНС — центральная нервная система
Болезнь Гоше (БГ) относится к группе лизосомных болезней накопления, характеризующихся патологическим отложением в органах и тканях промежуточных продуктов нормального метаболизма вследствие недостаточной активности лизосомных ферментов. При БГ нарушено ферментативное расщепление гликосфинголипидов в результате наследственного дефицита активности кислой β-глюкозидазы (глюкоцереброзидаза — ГЦБ) [1]. В редких случаях причиной БГ является наследственный дефицит белка — активатора ГЦБ сапозина С [2].
БГ — наиболее частая лизосомная болезнь накопления, встречается с частотой от 1:40 000 до 1:60 000 у представителей всех этнических групп; в популяции евреев ашкенази распространенность заболевания достигает 1:450—1:1000. Болезнь наследуется по аутосомно-рецессивному типу. Ген, кодирующий ГЦБ (ген GBA), локализуется в регионе q21 на 1-й хромосоме. Наличие 2 мутантных аллелей гена GBA (гомозиготное наследование) ассоциируется со снижением (или отсутствием) каталитической активности ГЦБ, что приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше) — перегруженных липидами макрофагов [1].
Накопление нерасщепленных продуктов метаболизма в цитоплазме макрофагов сопровождается продукцией этими клетками провоспалительных цитокинов, аутокринной стимуляцией моноцитопоэза и увеличением абсолютного количества макрофагов в местах их «физиологического дома» (селезенка, печень, костный мозг, легкие), что проявляется увеличением размеров селезенки и печени, инфильтрацией макрофагами костного мозга, легких и других органов [3, 4].
Основными клиническими проявлениями БГ служат спленомегалия, гепатомегалия, цитопения и поражение костей. В редких случаях у детей наблюдаются симптомы поражения центральной нервной системы (ЦНС), что сопряжено с крайне неблагоприятным прогнозом [1]. В соответствии с наличием или отсутствием неврологических проявлений выделяют 3 типа заболевания: I — наиболее частый клинический вариант, без неврологических проявлений; II (острый нейронопатический) — встречается у детей раннего возраста, характеризуется тяжелым поражением ЦНС, приводящим к летальному исходу в возрасте младше 2 лет; III (хронический нейронопатический) — симптомы поражения ЦНС могут проявляться как в раннем, так и в подростковом возрасте.
Поражение костно-суставной системы при БГ отличается выраженной гетерогенностью и варьирует от бессимптомной остеопении и колбообразной деформации дистальных отделов бедренных костей (колбы Эрленмейера) до тяжелейшего остеопороза и ишемических (асептических) остеонекрозов с развитием вторичных остеоартрозов и, как следствие, необратимых ортопедических дефектов. Именно поражение костно-суставной системы определяет тяжесть течения БГ и качество жизни пациентов. Причины столь ярко выраженной гетерогенности поражения костно-суставной системы не установлены.
Стандартом современной диагностики БГ является биохимическое определение активности ГЦБ в лейкоцитах крови (ферментная диагностика). Диагноз подтверждают при снижении активности фермента менее 30% от нормального значения. Степень снижения активности фермента не коррелирует с тяжестью течения заболевания [1].
Молекулярный анализ для выявления мутаций гена GBA позволяет верифицировать диагноз БГ, однако в настоящее время не является обязательным и используется для дифференциальной диагностики сложных клинических случаев или научного анализа [4].
Лечение пациентов с БГ заключается в назначении пожизненной заместительной ферментной терапии (ЗФТ) рекомбинантной глюкозидазой, дозы которой варьируют в зависимости от тяжести клинических проявлений и стадии лечения. Своевременное назначение ЗФТ позволяет остановить прогрессирование болезни и предотвратить необратимое поражение жизненно важных органов. Показания к назначению ЗФТ базируются исключительно на предшествующем эмпирическом опыте и включают детский возраст, выраженную органомегалию, глубокую цитопению, тяжелое поражение костей. Разработка ранних критериев неблагоприятного течения БГ остается актуальной клинической задачей.
Целью настоящего исследования явилась характеристика генотипов и генофенотипических корреляций у пациентов с БГ I типа в Российской Федерации.
Материалы и методы
Обследовали 100 взрослых пациентов с БГ I типа. Пациенты находились на обследовании и лечении в научно-клиническом отделении орфанных заболеваний Гематологического научного центр МЗ РФ в период с 2001 по 2012 г. Группу наблюдения составили 41 мужчина и 59 женщин в возрасте от 18 до 80 лет (средний возраст 31 год).
Диагноз БГ был установлен на основании характерной клинической и морфологической картины заболевания и во всех случаях подтвержден ферментной диагностикой — определением активности кислой β-глюкозидазы в лейкоцитах периферической крови. Ферментная диагностика осуществлялась в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра РАМН (зав. — к.м.н. Е.Ю. Захарова).
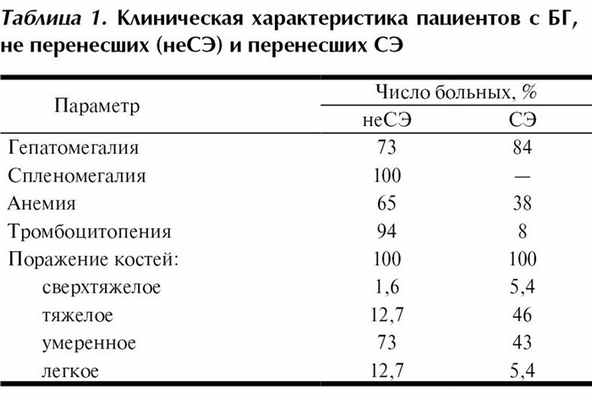
У 37 (37%) больных за 1—30 лет до поступления под наше наблюдение проведена диагностическая спленэктомия (СЭ). Клиническая и лабораторная картина БГ существенно отличается у больных, перенесших и не переносивших СЭ (табл. 1).
Тяжесть поражения костно-суставной системы оценивали по клиническим данным (наличие переломов, артрозов, других ортопедических дефектов) и результатам инструментальных исследований: рентгенографии и магнитно-резонансной томографии (МРТ) бедренных костей с захватом тазобедренных и коленных суставов. При необходимости проводили рентгенографию и МРТ других костей, а также денситометрию (двойную энергетическую рентгеновскую абсорбциометрию). На основании результатов инструментальных исследований определяли степень инфильтрации костей, а также наличие остеонекрозов, асептических некрозов головки или шейки бедренных костей, патологических переломов, вторичных остеоартрозов. На основании совокупности указанных данных больных разделили на 4 группы: с легким, умеренным, тяжелым и сверхтяжелым поражением костей.
Согласно данным, представленным в табл. 1, характерными клиническими проявлениями у больных без СЭ (n=63) были спленомегалия (у 100%), тромбоцитопения (94%) и вовлечение костей (100%), однако тяжелое и сверхтяжелое поражение костей выявили лишь у 8 (12,7%) и 1 (1,6%) больных соответственно.
В то же время у остальных 86% больных имелось легкое или умеренное поражение костно-суставной системы без необратимых ортопедических дефектов.
Больные, перенесшие СЭ (n=37), характеризовались наличием гепатомегалии, преимущественно нормальными показателями гемограммы (см. табл. 1). Вместе с тем тяжелое или сверхтяжелое поражение костей определялось у 46 и 5,4% пациентов соответственно, что согласуется с данными литературы, свидетельствующими о неблагоприятном течении БГ у больных, перенесших СЭ [1].
Скрининговое исследование для выявления 4 наиболее частых мутаций гена GBA (N370S, 84GG, L444P, IVS2+1) осуществляли методом аллель-специфической полимеразной цепной реакции в реальном времени. Исследование проводили в лаборатории молекулярной гематологии Гематологического научного центра (зав. — д.б.н. А.Б. Судариков). Праймеры и зонды разработаны А.Б. Судариковым и соавт. (данные не опубликованы).
Результаты и обсуждение
Повреждения гена GBA включают миссенс-мутации, сплайсинговые мутации, вставки, делеции нуклеотидов, кроссоверы между структурным геном и псевдогеном, конверсии генов (негомологичные рекомбинации) [5]. БГ I типа ассоциируется с миссенс-мутациями гена GBA и частичным дефицитом глюкозидазы. Напротив, инактивирующие точковые мутации, рекомбинантные аллели и внутригенные делеции сопряжены с накоплением продуктов деградации гликосфинголипидов в нервной ткани (нейроны, адвентициальные клетки, микроглия) и развитием нейронопатических типов БГ (II и III).
К настоящему времени описано около 350 различных мутаций гена GBA, из которых 4 (N370S, L444P, 84GG, IVS2+1) встречаются наиболее часто и составляют 90% всех мутантных аллелей гена GBA в популяции евреев ашкенази и около 60% мутантных аллелей у больных других этнических групп [5, 6].
Наиболее распространенная мутация гена GBA при БГ I типа — asn 370 → ser (N370S), которая вызвана заменой A на G в позиции 1226 комплементарной ДНК. Эта мутация имеется в 67,2% мутантных аллелей у евреев ашкенази и в 35% у пациентов других национальностей [7]. По данным Международного регистра, наиболее частым генотипом БГ в мире является генотип N370S/N370S [8].

В ходе молекулярно-генетических исследований, проведенных у российских пациентов с БГ, получены следующие результаты. Мутацию N370S выявили у 93 (93%) больных российской группы (Hetero — у 72, Homo — у 21), мутацию L444P hetero — у 21, мутацию IVS2+1 hetero — 2, мутацию 84GG hetero — у 1; в том числе генотип N370S/N370S — у 21, генотип N370S/L444P — у 18, генотип N370S/IVS2+1 — у 2, генотип N370S/? — у 52 пациентов (табл. 2).
Миссенс-мутация leu 444 → pro (L444P) вызвана заменой T на С в позиции 1448 комплементарной ДНК. Данная мутация встречается с частотой 3,1% мутантных аллелей в популяции евреев ашкенази и 27,5% аллелей у пациентов нееврейской национальности [7]. По данным Международного регистра БГ, генотип N370S/L444P занимает второе место по распространенности [8].
В российской группе мутацию L444P выявили у 21 больного, в том числе генотип N370S/L444P — у 18, L444P/84GG — у 1, L444P/? — у 2.
Мутации, приводящие к нарушению синтеза ГЦБ, в гомозиготном и сочетанном гетерозиготном (84GG/IVS2+1) состоянии являются летальными. Мутация 84GG приводит к сдвигу рамки считывания, что препятствует трансляции глюкозидазы, а мутация IVS2+1 (замена G на А в позиции 1067) нарушает сплайсинг первичного транскрипта вследствие удаления из него 2-го экзона [9].
Мутация 84GG в гетерозиготном состоянии встречается с частотой 12,5% мутантных аллелей в популяции евреев ашкенази и 0,25% — у пациентов других национальностей [7]. В нашей группе гетерозиготную мутацию 84GG выявили у 1 больного.
IVS2+1 в гетерозиготном состоянии встречается с частотой 3,1% мутантных аллелей в популяции евреев ашкенази и практически не встречается среди пациентов нееврейской национальности [7].
В российской группе мутацию IVS2+1 выявили у 2 больных в гетерозиготном состоянии в сочетании с мутацией N370S.
У 4 (4%) больных российской группы ни одной мутации не выявлено.

Результаты предшествующих клинических исследований не выявили корреляций между генотипом и фенотипом БГ [7]. Для выяснения прогностического значения генотипа БГ в российской популяции больных мы провели сравнительный анализ результатов молекулярно-генетических исследований и клинических данных, характеризующих степень тяжести поражения костно-суставной системы (табл. 3). Согласно приведенным данным анализ не выявил очевидной связи между генотипом БГ и тяжестью поражения костно-суставной системы. Генотип N370S/? имелся у больных с легким, умеренным, тяжелым и сверхтяжелым поражением костей примерно с равной частотой. Однако отсутствие информации о втором мутантном аллеле пока не позволяет сделать окончательное заключение.
Заключение
Результаты проведенных клинических и молекулярно-генетических исследований позволили выявить следующее: у 93% российских пациентов с БГ I типа имеется мутация N370S; у 52% больных имеется генотип N370S/?, где второй аллель представлен мутацией, не входящей в число 4 наиболее частых мутаций гена GBA; отсутствует очевидная корреляция между типом мутации гена GBA и тяжестью поражения костно-суставной системы; СЭ приводит к регрессу цитопении, но ассоциируется с последующим тяжелым поражением костно-суставной системы у 51% больных, перенесших СЭ.
В соответствии с этим больным с неясной цитопенией и спленомегалией СЭ следует проводить после исключения диагноза БГ. Пациенты с БГ, подвергавшиеся СЭ, нуждаются в неотложном назначении ЗФТ для предотвращения тяжелого поражения костно-суставной системы и развития необратимых ортопедических дефектов.
Читайте также: