Болезнь Вильсона Коновалова: причины, симптомы и лечение
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Болезнь Вестфаля-Вильсона-Коновалова (БВВК) — наследственное заболевание, при котором происходит нарушение обмена меди. Болезнь протекает с преимущественным поражением печени, почек, центральной нервной системы (ЦНС).
Вследствие генетического дефекта в 13 хромосоме происходит увеличение всасывания меди в кишке, снижение синтеза белка церулоплазмина и выделения меди с желчью (при этом, увеличено содержание токсичной «свободной» меди в крови и тканях, накопление ее и поражение в печени, почках, мозге, роговице).
Повреждение печени характеризуется этапным развитием ожирения печени (стеатоз), разрушения клеток печени (некроз), соединительной ткани в печени (фиброз) и его крайней степенью выраженности — цирроз печени.
Симптомы
Выделяют бессимптомную (выявляется только по лабораторным изменениям крови), брюшную (абдоминальную — с преимущественным поражением печени), церебральную (с преимущественным поражением головного мозга) и смешанные формы заболевания.
Симптомы можно условно разделить на внепеченочные (затрагивающие другие органы) и печеночные.
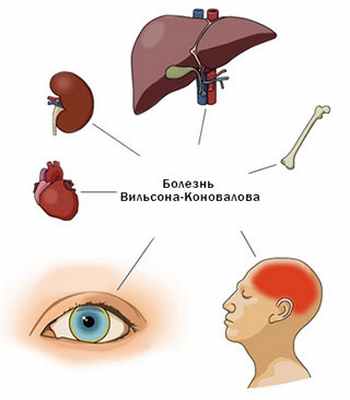
Внепеченочные проявления
Включают поражения почек, нервной системы (дрожание конечностей, нарушение речи, повышенное слюноотделение и пр.), психозы, поражение глаз (кольца Кайзера-Флейшера), сердца, эндокринной системы, системы крови (повышенное разрушение эритроцитов), костей (патологические переломы), кожи.
Печеночные проявления болезни
Являются наиболее частыми и отмечаются у 42% больных.
- Бессимптомные формы могут характеризоваться повышением активности ферментов печени (АЛТ, АСТ) в крови или увеличением печени или селезенки.
- Острый гепатит — часто развивается в детском возрасте, у 25% больных — желтуха, отсутствие аппетита, слабость, увеличение печени, изменение биохимических показателей. Часто проходит самостоятельно, но временно.
- Хронический гепатит часто развивается в возрасте 11–25–30 лет.
- Цирроз печени (выявляется у всех больных с поражением нервной системы)
- Фульминантная (молниеносная) форма печеночной недостаточности может развиться на фоне хронического гепатита или, чаще, цирроза печени.
Внимание!
Не откладывайте посещение врача-гепатолога, если у вас:
- выявляются изменения в биохимических показателях функции печени (АсАТ, АлАТ), а причина неизвестна
- имеется сочетание изменений функции печени и поражение нервной системы (нарушение почерка, дрожание рук, непроизвольные подергивания, затруднение речи, изменение голоса и пр.)
- если подобные изменения также обнаружены у родных братьев и сестер или родители являются родственниками — вы являетесь ребенком в близкородственном браке.
Диагностика
Главное в лечении болезни Вестфаля-Вильсона-Коновалова — это своевременное и правильное диагностирование этого заболевания!
Диагностика БВВК начинается с консультации гепатолога, на которой врач собирает анамнез наследственности (близкородственные браки, заболевания печени и нервной системы в семье и пр.).
Врач назначает необходимый спектр лабораторно-инструментальной диагностики, соответствующий мировым стандартам современной гепатологии.
Инструментальные исследования:
- — оценкой фиброза печени
- МРТ головного мозга
- по показаниям — биопсия печени.
Анализы:
- клинический анализ крови
- биохимическое исследование крови с оценкой показателей функции печени
- исследование обмена меди в крови и моче, определение в крови концентрации белка церулоплазмина, транспортирующего медь
- генетическое исследование крови на типичные мутации или углубленное обследование на редкие мутации гена
- определяются стадия фиброза, степень воспаления в печени по показателям крови в виде ФиброТеста/ФиброМакса.
Лечение
Лечение нарушения обмена меди в гастро-гепатоцентре ЭКСПЕРТ включает в себя:
- обязательное совместное ведение пациента врачом-гепатологом и врачом-неврологом
- индивидуальный подбор и применение препаратов, выводящих медь из организма
- изменение питания с помощью врача-диетолога с исключением продуктов, богатых медью: баранина, мясо птиц, печень, колбасные изделия, ракообразные, грибы, салат, щавель, лук-порей, редис, бобовые, орехи, каштаны, перец, чернослив, мед, шоколад, какао
- применение гепатопротекторов, антиоксидантов, препаратов, улучшающих состояние сосудистой стенки по индивидуально разработанным схемам
- проведение терапии, направленной на обратное развитие фиброза и цирроза печени
- при необходимости, в тяжелых случаях, подготовка пациентов для представления на отборочную комиссию по трансплантации печени.
Лечение нарушения обмена меди предъявляет особые требования к контролю состояния вашего здоровья, полнотой выполняемых назначений и рекомендаций. Именно поэтому на протяжении всего лечения необходимо регулярно посещать врача-гепатолога, врача-невролога согласно разработанному плану.
Если после консультации у вас возникают дополнительные вопросы, вы всегда можете обратиться к врачу-куратору за любыми разъяснениями.
Прогноз
Прогноз при болезни Вестфаля-Вильсона-Коновалова связан со степенью нарушения печеночных функций, тяжестью поражения нервной системы, а также зависит от желания пациента следовать рекомендациям врача и его дисциплинированности.
БВВК является прогрессирующим заболеванием и при отсутствии своевременной терапии есть высокий риск смерти от осложнений цирроза печени или (реже) от прогрессирующей неврологической симптоматики.
- Неблагоприятный прогноз — при острой неврологической форме, развитии осложнений цирроза печени.
- Нормализация печеночных функций происходит на 1–2 году терапии и не прогрессирует при полном выполнении всех рекомендаций.
- Оперативное вмешательство необходимо при фульминантном (молниеносном) течении заболевания.
- Неврологическая симптоматика БВВК частично обратима при адекватной терапии и проведении трансплантации печени, что связано с необратимыми поражениями структур головного мозга токсическими концентрациями меди.
Когда удается достичь ремиссии, врач-гепатолог назначает итоговое обследование ЦНС, печени и других органов и систем. Также врач совместно с вами планирует сроки контрольных посещений и обследования с интервалом 1 раз в 3 месяца в течение года, и 2 раза в год – пожизненно.
Болезнь Вильсона-Коновалова
Болезнь Вильсона–Коновалова (гепато-лентикулярная дегенерация) относится к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов. Если своевременно не начать лечение, направленное на выведение токсичных избытков меди из организма, то через 5–7 лет больной обречен на смерть. Болезнь поражает 25% братьев и сестер в семье при клинически здоровых родителях, которые являются носителями аномального гена (аутосомно-рецессивный тип наследования). Заболевают только те индивидуумы, которые унаследовали два мутантных гена, то есть по одному от матери и от отца – гомозиготные носители мутации; лица, которые от одного из родителей получили мутантный ген, а от другого – нормальный ген, являются гетерозиготными носителями мутации и остаются здоровыми.
Открытый недавно ген болезни отвечает за синтез медь-транспортирующего белка (АТР7В). При гепатолентикулярной дегенерации обмен меди и медьсодержащих белков нарушается, появляется избыток “свободной” меди, которая в больших количествах откладывается в печени, мозге, роговице, а также выделяется с мочой. Не случайно диагностика болезни базируется на обнаружении характерных нарушений медного обмена. Благодаря идентификации гена в настоящее время возможна и ДНК- диагностика этого заболевания.
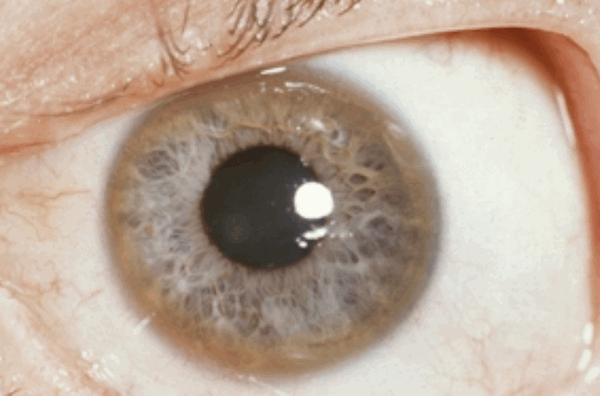
Поражение печени избытком “свободной” меди проявляется циррозом печени. Поражение мозга приводит к развитию тяжелой неврологической симптоматики: дрожанию конечностей и всего туловища, повышению мышечного тонуса, иногда сопровождающемуся болезненными спазмами, нарушением речи, глотания, снижению интеллекта. Отложение меди в роговице (по краю радужной оболочки) обусловливает формирование кольца Кайзера–Флейшера – буро-зеленоватого пигмента. По этому признаку диагноз болезни можно поставить безошибочно.
Гепато-лентикулярная дегенерация известна с глубокой древности. Дошедшее до нас изображение египетского фараона Тутанхамона, по мнению крупнейшего специалиста J. Walshe, не исключает вероятности, что он страдал этим заболеванием. Институт неврологии РАМН в течение многих лет занимается проблемой гепато-лентикуляной дегенерации. Знаменитый отечественный невролог академик АМН СССР Н.В. Коновалов, один из основателей Института, посвятил этому заболеванию две монографии, последняя из которых в 1964 году была удостоена Ленинской премии. В последующие годы данное заболевание продолжало успешно изучаться сотрудниками нейрогенетического отделения Института неврологии, под наблюдением которых за 40 лет находилось свыше500 семей, отягощенных этим недугом. Весь многолетний опыт Института свидетельствует о том, что ключевой проблемой является ранняя диагностика гепатолентикулярной дегенерации. Чем раньше начать лечение (в идеале – еще на досимптоматической стадии либо на доневрологическом этапе, то есть до появления признаков поражения мозга), тем лучше эффект. Вот почему, если в семье есть хоть один ребенок, страдающий этим заболеванием, необходимо тщательное обследование всех его братьев и сестер, в том числе с использованием самых современных биохимических и молекулярно-генетических методов.
Заподозрить раннюю стадию болезни можно на основании следующих признаков: перенесенной желтухи; повторных кровотечений из носа, кровоточивости десен либо множественных кровоподтеков; сосудистых “звездочек” на коже груди и спины; своеобразных “полосок” (белых, меняющих периодически окраску на красновато-синюшную) на бедрах и в подмышечных областях; гормональных нарушений в виде аменореи или дисменореи у девушек, гинекомастии (нагрубание грудных сосков) у юношей, а также акромегалии(увеличение носа, подбородка, утолщение губ); снижения интеллекта и изменений психики в виде чередования дурашливости и пониженного настроения, трудностей усвоения нового материала, проблем с успеваемостью в школе.
Гепато-лентикулярная дегенерация может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко – в 50–60 лет. Чем раньше начинается заболевание, тем тяжелее оно протекает (при отсутствии лечения). Однако болезнь Вильсона–Коновалова – редкий пример наследственного нарушения, для которого разработаны высокоэффективные методы лечения: даже при появлении тяжелой неврологической симптоматики систематическое лечение обычно дает “драматический” эффект, вплоть до исчезновения всех симптомов или резкого их уменьшения. Пациенты вновь могут полностью обслуживать себя, вести домашнюю работу, учиться, работать по профессии, создать семью и родить здорового ребенка (под нашим наблюдением находятся 30 женщин, страдающих гепатолентикулярной дегенерацией и благополучно родивших здоровых детей). Пациентам с гепатолентикулярной дегенерацией необходимо регулярно наблюдаться у постоянного лечащего врача.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.
Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности.
При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). Нами было предложено комбинированное лечение D-пеницилламином и препаратами цинка, что дает возможность снизить дозу и избежать побочных явлений. У больных в пресимптоматической стадии достаточно лечения только препаратами цинка.
В настоящее время за рубежом в тяжелых случаях болезни, не поддающихся консервативному лечению, широко применяется пересадка печени. При удачном исходе операции больной полностью выздоравливает и не нуждается в дальнейшем приеме препаратов. В России делаются первые шаги в этом направлении, и одним из таких шагов является разработанный нами совместно с Институтом трансплантологии и искусственных органов метод био-гемоперфузии с изолированными живыми клетками печени и селезенки – так называемый аппарат “вспомогательная печень”.
Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени.
Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики.
Болезнь Вильсона — Коновалова
Болезнь Вильсона — Коновалова (гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — редкое наследственное заболевание, нарушение метаболизма меди, характеризующееся ее избыточным накоплением в тканях и жизненно-важных органах (в основном в печени, почках, головном мозге, глазах).
Распространенность болезни Вильсона — Коновалова во всем мире составляет 1 случай на 30 000–100 000 человек. Данные приблизительные, поскольку пациенты с гепатолентикулярной дегенерацией иногда получают ошибочный диагноз (в этот список входят различные болезни печени, неврологические заболевания и психические расстройства). Оценка носительства гена заболевания колеблется, однако считается, что его носителем может быть 1 человек из 90.
Причины возникновения
Болезнь Вильсона — Коновалова наследуется по аутосомно-рецессивному типу (мутации гена — в данном случае ATP7B — наследуются от обоих родителей). При таком типе наследования у каждого ребенка носителей дефектного гена существует 25% вероятность заболеть и 50% вероятность носительства мутации.
Ген ATP7B отвечает за метаболизм меди и играет важную роль в выведении избытка меди из организма. На сегодняшний день выявлено более 300 мутаций гена ATP7B.
Симптомы
Возраст манифестации заболевания варьируется: симптомы могут начать проявляться у ребенка 3–5 лет или же болезнь не дает о себе знать десятилетиями. Однако чаще всего болезнь Вильсона — Коновалова развивается к сорока годам, в редких случаях — после пятидесяти лет. Клиническая картина заболевания зависит от пола и возраста, так, у детей (средний возраст — 10 лет) чаще преобладают поражения печени.
К признакам и симптомам заболевания относятся:
- проявления со стороны печени (гепатомегалия, острый гепатит, хронический гепатит, цирроз, фульминантная печеночная недостаточность) обычно предшествуют неврологическим и психиатрическим симптомам;
- изменение цвета кожи, слизистых оболочек и склер глаз на желтый; и живота (асцит);
- боль в верхней правой части живота;
- возникновение синяков на коже, коагулопатия;
- повышенная утомляемость;
- потеря аппетита и веса; и рвота;
- неврологические симптомы (тремор, нарушение координации и походки, дисфагия, дизартрия, хорея, спастичность, дистонические позы, мышечная ригидность);
- психические расстройства (эмоциональная нестабильность, фобии, тревожность, депрессия, компульсивное поведение, агрессивность, изменения личности и поведения) широко варьируются от пациента к пациенту, проявляются одновременно с неврологическими симптомами или развиваются в течение примерно 3 лет после их старта;
- кольца Кайзера — Флейшера на границе роговицы глаз (почти у всех пациентов с неврологическими симптомами и психическими расстройствами);
- аменорея;
- задержка полового созревания;
- выкидыши, бесплодие;
- артралгия, боль в костях;
- поражения костей и суставов (остеопороз, остеофиты);
- кардиомиопатия;
- гемолитическая анемия;
- гематурия;
- нефротический синдром;
- почечный канальцевый ацидоз;
- камни в почках;
- гепатоцеллюлярная карцинома (редко).
Диагностика
Раннее выявление болезни Вильсона — Коновалова и своевременное лечение позволяют сохранить нормальное качество жизни пациента и предотвратить опасные осложнения. Диагноз устанавливается на основании физикального осмотра, сбора жалоб, личного и семейного анамнеза пациента, лабораторной диагностики (проводятся биохимические анализы крови и мочи, которые могут показать сниженную концентрацию медь-содержащего белка церулоплазмина в плазме крови, повышение уровня печеночных ферментов, тромбоцитопению, повышенное содержание меди в моче), осмотра глаз офтальмологом с помощью щелевой лампы, КТ или МРТ головного мозга (у пациентов с неврологическими симптомами). Если лабораторные исследования не подтверждают и не исключают диагноз, врач может назначить биопсию печени, а также молекулярно-генетическое тестирование.
Болезнь Вильсона — Коновалова дифференцируют (различают) со следующими заболеваниями: вирусный гепатит, аутоиммунный гепатит, неалкогольная жировая болезнь печени, алкогольный цирроз печени, первичный склерозирующий холангит, первичный билиарный цирроз, неврологическими патологиями (эссенциальный тремор, болезнь Паркинсона с ранним началом, дистония, болезнь Хантингтона).
Лечение болезни Вильсона — Коновалова
Лекарства, воздействующего на причину болезни Вильсона — Коновалова, не существует. Пожизненное лечение направлено на снижение уровня меди в организме до нормального (нетоксичного), снижение прогрессирования заболевания и уменьшение симптомов, которые с ним связаны. Оно включает хелатирующие агенты (D-пеницилламин или триентин), которые связывают и выводят медь из организма с мочой; ацетат цинка, предотвращающий кишечную абсорбцию (всасывание) меди. Дополнительно врач может порекомендовать соблюдение диеты, исключающей продукты с повышенным содержанием меди (печень, шоколад, грибы, моллюски, орехи). Проводится динамическое наблюдение (пациент периодически сдает анализы крови и мочи) с целью оценки эффективности медикаментозной терапии.
Во время беременности лечение заболевания рекомендуется не прекращать, в этот период врач может снизить дозировку хелатирующего агента, прием ацетата цинка продолжается по стандартной схеме. Кормление грудью при использовании хелатирующего агента исключается, данных по ацетату цинка недостаточно.
При острой печеночной недостаточности, вызванной прогрессированием болезни Вильсона — Коновалова, необходима трансплантация печени.
Особенности и преимущества лечения болезни Вильсона — Коновалова в клинике Рассвет
Врачи клиники Рассвет специализируются на диагностике и лечении редких синдромов и болезней, в область их интересов в том числе входит помощь пациентам с гепатолентикулярной дегенерацией (болезнью Вильсона — Коновалова).
Наши гепатологи — врачи высокой квалификации, прекрасно подготовлены и имеют большой практический опыт. Любое редкое заболевание требует мультидисциплинарного подхода, поэтому пациент с болезнью Вильсона — Коновалова при необходимости направляется на консультацию к офтальмологу, нефрологу, психотерапевту, психиатру.
Пациентам с подозрением на болезнь Вильсона — Коновалова проводятся все необходимые диагностические исследования для подтверждения или исключения диагноза, индивидуально подбирается схема терапии.
Невролог «СМ-Клиника» рассказала о течении болезни Вильсона-Коновалова у взрослых
В 1912 году одновременно в нашей стране и за рубежом была описана особая наследственная патология, которая получила свое название по авторам — болезнь Вильсона-Коновалова. Это наследственная болезнь и она опасна. Можно ли ее вылечить — выясним с экспертом.

АЛЕНА ПАРЕЦКАЯ
Врач-патофизиолог, иммунолог, член
Санкт-Петербургского общества патофизиологов
ВАЛЕНТИНА КУЗЬМИНА
К.м.н., врач-невролог «СМ-Клиника»
Один из самых характерных признаков болезни – это патологическое накопление меди в области различных органов, повреждение тканей, особенно – печени, проблемы нервной системы, изменения в радужке глаза.
Что нужно знать о болезни Вильсона-Коновалова
- Причины
- Симптомы
- Лечение
- Профилактика
- Вопросы и ответы
Что такое болезнь Вильсона-Коновалова
Термином болезнь Вильсона-Коновалова называют наследственную патологию. Она возникает, когда родители передают ребенку дефектный ген (АТР7В). Состояние относится к аутосомно-рецессивным патологиям, то есть возникает, если каждый из родителей несет подобный ген в своих клетках и ребенок наследует сразу оба гена – от матери и от отца.
Этот дефектный ген дает команды к синтезу белка, который регулирует обмен и перенос меди внутри организма. При его дефекте медь копится в печени, концентрируется в нервных ганглиях, откладывается в радужке глаза. Патология встречается нечасто, ее иногда очень сложно распознать, особенно, если в семье нет подобных больных.
Причины болезни Вильсона-Коновалова у взрослых
Ключевой процесс при этой патологии – наследование дефектного гена от родителей. Он располагается в 13-ой хромосоме и регулирует обмен меди.
В среднем, организм взрослых людей содержит приблизительно 50 - 70 мг меди и в сутки ему нужно не более 2 мг элемента, который поступает с пищей.
Подавляющий объем микроэлемента (95%) переносится в тесной связке с белком плазмы – церулоплазмином. Его постоянно образует печень, и только около 5% меди переносится вместе с альбумином.
Медь нужна для участия в метаболических процессах, в том числе – окислительных. Если развивается болезнь Вильсона, выведение ее нарушается, повышается концентрация в плазме, оттуда она разносится в ткани. Основное накопление меди происходит в мозге, в области радужки, внутри печени, а также в почках. Избыток микроэлемента оказывает токсический эффект.
Симптомы болезни Вильсона-Коновалова у взрослых
Возможные проявления очень разнообразны. Чаще всего страдает печень (около 40 - 50% случаев), а в остальных случаях это могут отмечаться неврологические поражения и проблемы психики. При поражении нервной системы и зрения появляется типичный симптом – проявление кольца Кайзера-Флейшера (возникает за счет отложения меди в радужке со специфическим ее бурым окрашиванием).
При брюшной форме болезни симптомы обычно проявляются ближе к 40 годам. Среди ключевых признаков:
- цирроз печени;
- хронический или фульминантный (молниеносный) гепатит.
Вариант дрожательной болезни Вильсона обычно возникает в возрастном промежутке от 10 лет до 30 - 35 лет. Могут возникать такие проявления как дрожание, замедление движений, заторможенность речи, приступы эпилепсии, проблемы психики.
Самая редкая форма болезни – это экстрапирамидно-корковые расстройства. Он похожи на все формы, дополнительно будут судорожные приступы, выраженные проблемы интеллекта, расстройства движений.
Лечение болезни Вильсона-Коновалова у взрослых
Чтобы проводить эффективное лечение, важно как можно ранее установить диагноз. Это не всегда просто, особенно в ситуациях, когда нет типичных симптомов и поражения радужки глаза с появлением кольца. Чаще всего пациенты приходят к неврологу, гастроэнтерологу или проблема выявляется у окулиста.
Диагностика
Если речь идет о проявлении глазных симптомов, врач предварительно осматривает состояние глаз щелевой лампой для того, чтобы подтвердить наличие кольца Кайзера-Флейшера.
Показано назначение биохимических тестов крови и мочи, которые покажут повышенное содержание меди в моче и сниженную концентрацию церулоплазмина в плазме крови.
КТ или МРТ покажут атрофические процессы в области мозга и мозжечка, повреждение базальных ядер.
Дополнительно проводится консультация генетика и ряд генетических тестов, выявляющих дефектные гены.
Современные методы лечения
Основной метод лечения при этой болезни – назначение тиоловых препаратов, особенно – унитиола либо D-пеницилламина, купренила. Препараты принимают длительно, врач подбирает наиболее оптимальную дозу, которая позволит избежать побочных эффектов.
Дополнительно врач может применять препараты из группы нейролептиков, при ригидности мышц – леводопу или карбидопу.
При тяжелом течении показана трансплантация печени, иммунсупрессивная терапия. Возможно применение биогемоперфузии с изолятом живых клеточных элементов селезенки с печенью.
Дополнительно необходимо соблюдение диеты с исключением продуктов, содержащих большое количество меди.
Профилактика болезни Вильсона-Коновалова у взрослых в домашних условиях
– Для профилактики патологии, – говорит врач-невролог Валентина Кузьмина, – необходимо придерживаться диеты №5, а также ограничить потребление меди до 1 г в сутки – исключить орехи, сухофрукты, шоколад, раков, печенье, цельную пшеницу. Также рекомендован прием препаратов группы витамина В6, унитиола, триентина.
Популярные вопросы и ответы
Мы поговорили о проблемах болезни Вильсона-Коновалова, ее осложнениях и возможности самолечения с врачом-неврологом Валентиной Кузьминой.
Какие могут последствия при болезни Вильсона-Коновалова?
Среди основных последствий болезни Вильсона-Коновалова можно выделить:
- поражение печени, особенно, если развивается цирроз печени;
- психические заболевания – существенное замедление умственного развития, психозы;
- неврологические заболевания – нарушение координации, при котором есть еще и дрожание конечностей, расстройства ходьбы, повышенное слюноотделение.
Вызвать врача на дом необходимо при нарушении речи (дизартрия) и глотания (дисфагия), насильственном непроизвольном смехе или плаче, нарушении эмоционального состояния, умеренном снижении интеллекта.
Можно ли вылечить болезнь Вильсона-Коновалова народными средствами?
Нет, лечить болезнь Вильсона-Коновалова народными средствами ни в коем случае нельзя. Это только навредит и ухудшит проблемы печени и нервной системы. Обязательно обратитесь к специалисту.
Болезнь Вильсона
(Болезнь Вильсона; наследственная медная интоксикация)
, MD, PhD, University of Arkansas for Medical Sciences
- Патофизиология
- Клинические проявления
- Диагностика
- Прогноз
- Лечение
- Основные положения
Болезнь Вильсона приводит к накоплению меди в печени и других органах. Развиваются печеночные или неврологические симптомы. Диагноз основан на низком уровне в сыворотке церулоплазмина, высоком уровне экскреции меди с мочой, иногда результатах биопсии печени. Лечение заключается в диете с низким содержанием меди и лекарственной терапии, например, пеницилламином или триентином.
Болезнь Вильсона - нарушение обмена меди, развивающееся и у мужчин, и у женщин; распространенность 1 случай на 30000 человек. Люди, пораженные этой болезнью, гомозиготны по рецессивному мутантному гену, расположенному в 13-й хромосоме. У гетерозиготных носителей, составляющих приблизительно 1,1% населения, болезнь не проявляется.
Патофизиология болезни Вильсона
Генетический дефект при болезни Вильсона нарушает перенос меди. Нарушение переноса уменьшает секрецию меди в желчь, что вызывает перегрузку медью и, как следствие, ее накопление в печени, которое начинается с рождения. Нарушение переноса также препятствует включению меди в медьсодержащий белок церулоплазмин, что ведет к снижению сывороточного уровня церулоплазмина.
Развивается печеночный фиброз, в конечном итоге приводит к циррозу. Медь диффундирует из печени в кровь, а затем в другие ткани. Это приводит, главным образом, к деструктивным поражениям головного мозга, но также поражаются почки, репродуктивные органы и развивается гемолитическая анемия. Некоторое количество меди оседает по краю роговицы и радужной оболочки, вызывая появление колец Кайзера-Флейшера. При осмотре видно, как кольца окружают радужную оболочку.
Симптомы и признаки болезни Вильсона
Симптомы болезни Вильсона обычно развиваются в возрасте от 5 до 35 лет, но могут также развиваться в диапазоне 2-72 лет.
Почти у половины пациентов, особенно подростков, первый симптом –
Гепатит - острый, хронический, активный, либо молниеносный
Но гепатит может развиться в любое время.
Приблизительно у 40% пациентов, особенно в молодом возрасте, первые симптомы отражают
Поражение центральной нервной системы (ЦНС)
Типичны двигательные нарушения, включая любую комбинацию тремора, дистонии, дизартрии, дисфагии, хореи, слюнотечения и нарушения координации. Иногда поражение ЦНС проявляется поведенческими или когнитивными отклонениями.
У 5–10% пациентов первый симптом это случайно замеченные золотые или зеленовато-золотые кольца Кайзера – Флейшера или полумесяцы (из-за отложения меди в роговице), аменорея, повторные самопроизвольные аборты, гематурии.
Диагностика болезни Вильсона
Обследование со щелевой лампой для выявления колец Кайзера-Флейшера
Сывороточный церулоплазмин, иногда сывороточная концентрация меди и 24-часовая экскреция меди с мочой
Иногда подтверждение провокационным тестом пеницилламином или биопсией печени
Болезни Вильсона следует подозревать у человека младше 40 лет в любом из следующих случаев:
печеночная, неврологическая или психиатрическая патология неизвестной этиологии;
постоянное повышение печеночной трансаминазы неизвестной этиологии;
брат, сестра, родитель или кузен страдает болезнью Вильсона;
При подозрении на болезнь Вильсона необходимо провести исследование щелевой лампой для выявления колец Кайзера–Флейшера, измерить уровень церулоплазмина в сыворотке крови и экскрецию меди в суточной моче. Можно также измерить сывороточную концентрацию меди, но обычно достаточно определения сывороточного церулоплазмина. Также частое измерение уровней трансаминазы; высокие уровни соответствуют диагнозу.
Кольца Кайзера – Флейшера
Эти кольца в сочетании с двигательными неврологическими нарушениями или снижением уровня церулоплазмина патогномоничны для болезни Вильсона. Изредка кольца встречаются при других заболеваниях печени (например, желчной атрезии, первичном билиарном циррозе), но уровни церулоплазмина остаются неизменными.
Церулоплазмин
Уровень церулоплазмина в сыворотке крови (норма 20–35 мг/дл [200–350 мг/л]) при болезни Вильсона обычно снижен, но может быть и в норме. Он может быть также низким, особенно у гетерозиготных носителей и лиц с иными заболеваниями печени (например, вирусном гепатите, лекарственной или алкогольной болезни печени). Низкий уровень церулоплазмина у больных с кольцами Кайзера – Флейшера позволяет диагностировать заболевание. Кроме того, уровень 5 мг/дл (
Сывороточная медь
Иногда измеряют сывороточные уровни меди; однако, они могут быть высокими, нормальными или низкими.
Экскреция меди с мочой
При болезни Вильсона экскреция меди в суточной моче (норма ≤ 30 мкг/день) достигает > 100 мкг/день. Если сывороточный церулоплазмин низкий, а экскреция меди с мочой высокая, то диагноз ясен. Если уровни сомнительны, то диагноз можно подтвердить измерением экскреции меди с мочой после приема пеницилламина (пеницилламиновый провокационный тест); этот тест обычно не проводится у взрослых, потому предельные значения точно не установлены.
Биопсия печени
В сомнительных случаях (например, при повышении уровня трансаминазы, отсутствии колец Кайзера – Флейшера, неопределенных значениях церулоплазмина и меди в моче), диагноз ставится на основании биопсии печени для измерения концентрации меди в печени. Однако могут быть ложноотрицательные результаты из-за ошибки выборочного исследования (концентрация меди в печени очень сильно варьируется) или фульминантного гепатита (вызывает некроз, что приводит к высвобождению больших количеств меди).
Обследование для выявления болезни Вильсона
Поскольку раннее лечение наиболее эффективно, всем братьям и сестрам, кузенам и родителям пациента с болезнью Вильсона проводят скрининговое обследование для ее выявления. Скрининг включает обследование с помощью щелевой лампы и измерение уровней трансаминазы, сывороточных уровней меди и церулоплазмина, а также экскреции меди в суточной моче. Если какие-нибудь результаты являются патологическими, берется биопсия печени для того, чтобы измерить концентрацию меди в печени.
Младенцы не должны тестироваться до возраста 1 года, потому что уровни церулоплазмина низкие в течение первых нескольких месяцев жизни. Дети младше 6 лет с нормальными результатами тестов должны быть повторно протестированы спустя 5–10 лет.
Генетическое тестирование неосуществимо.
Прогноз при болезни Вильсона
Прогноз при болезни Вильсона обычно благоприятный, если болезнь до начала лечения не прогрессировала до поздних стадий.
Болезнь Вильсона без терапии приводит к фатальному исходу, и пациенты обычно погибают в возрасте до 30 лет.
Лечение болезни Вильсона (Overview of Wilson Disease)
Пеницилламин или триентин
Диета с низким содержанием меди
Для поддержки назначают пожизненное лечение низкими дозами пеницилламина или триентинина, или прием препаратов цинка внутрь
Непрерывное пожизненное лечение болезни Вильсона проводят обязательно, независимо от наличия симптомов. Накопление меди можно предупредить диетой с низким содержанием содержащих медь пищевых продуктов (например, следует избегать говяжьей печени, орехов кешью, спаржевой фасоли, овощных соков, моллюсков, грибов и какао) или лечением пеницилламином, триентином, а иногда - препаратами цинка перорально. Следует проверить содержание меди в питьевой воде, и рекомендовать пациенту не принимать витаминных и минеральных добавок, содержащих медь.
Пеницилламин является чаще всего используемым хелатирующим препаратом, но он имеет значительную токсичность (например, лихорадка, сыпь, нейтропения, тромбоцитопения, протеинурия). У людей с аллергией на пенициллин может возникнуть перекрестная реактивность. Пациентам в возрасте старше 5 лет его назначают перорально в дозе от 62,5 мг каждые 6 часов до 250 мг каждые 12 часов (250–500 мг/день 2–4 приема) и медленно повышают до максимальной дозы, от 250 мг каждые 6 часов до 750 мг каждые 12 часов (1000-1500 мг/день 2-4 приема). Маленьким детям назначают 10 мг/кг 2 раза в день или 6,7 мг/кг 3 раза в день(20 мг/кг/день) перорально. Пиридоксин 25 мг перорально назначается 1 раз/день в сочетании с пеницилламином. Иногда прием пеницилламина может вызвать ухудшение неврологических симптомов.
Препарат триентин гидрохлорид – также хелатирующее средство, является альтернативой пеницилламину. Он назначается в дозе 375–750 мг перорально 2 раза в день, или 250–500 мг 3 раза в день (750–1 500 мг/день).
Пероральный прием ацетата цинка в дозе 50 мг 3 раза в день может уменьшить всасываемость кишечником меди и тем самым предотвратить повторное накопление меди у больных с непереносимостью пеницилламина или триентина, или имеющих неврологические симптомы, не купируемые другими препаратами. (ВНИМАНИЕ: Пеницилламин или триентин не следует принимать одновременно с цинком, так как любой из этих лекарственных препаратов может связывать цинк, образуя соединение, не обладающее терапевтическим эффектом.)
Длительная лекарственная терапия обычно плохо соблюдается. После 1–5 лет нижнюю поддерживающую дозу лекарственной терапии следует пересмотреть. Рекомендуется регулярно последующее наблюдение у специалиста по заболеваниям печени.
Трансплантация печени при болезни Вильсона, осложнившейся фульминантным гепатитом или тяжелой печеночной недостаточностью, резистентной к лекарствам, может спасти пациенту жизнь.
Основные положения
Болезнь Вильсона - редкое аутосомно-рецессивное заболевание, при котором в различных органах накапливается медь.
Болезнь проявляется в детстве или в зрелом возрасте, как правило, от 5 до 35 лет.
Заболевание выявляют у людей с семейным анамнезом заболевания или при не имеющих явной причины поражениях печени, неврологических и психических нарушениях (в том числе при повышенном уровне трансаминаз).
Диагноз подтверждается исследованием с помощью щелевой лампы при выявлении колец Кайзера–Флейшера и при низком уровне церулоплазмина и высоком уровне экскреции меди в суточной моче.
Больным следует рекомендовать диету с низким содержанием меди и лечить их пеницилламином, триентином или, если эти препараты неэффективны или непереносимы, пероральным приемом цинка.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: