Морфология и наследственность синдрома Альпорта
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
О работе отделения наследственных и приобретенных болезней почек Научно-исследовательского клинического института педиатрии имени академика Ю.Е. Вельтищева ФГБОУ ВО РНИМУ им. Н.И. Пирогова рассказывает его главный научный сотрудник, доктор медицинских наук, профессор Лариса ПРИХОДИНА.
Лариса Серафимовна, о вашем отделении ходят легенды. Как по-вашему, почему?
Потому что здесь работают настоящие фанаты своего дела, профессионалы с большой буквы. Его основателем был академик Юрий Евгеньевич Вельтищев, чье имя ныне носит наш Институт, а первым руководителем – профессор Майя Сергеевна Игнатова>, одна из пионеров мировой педиатрической нефрологии, ставшая одним из инициаторов создания Европейского общества педиатров-нефрологов .
Попасть на работу в этот отдел в 1996-м году было для меня большой удачей. Ведь в институте мы изучали нефрологию всего лишь 10 дней. Для меня и многих моих коллег работа в этом отделении стала настоящими университетами. Благодаря нашему учителю и нашим пациентам, которые постоянно заставляют нас думать и искать. Искать ответы на многие вопросы, которые ставит перед нами их редкая болезнь.
Каков спектр проблем, которыми вы занимаетесь?
>Он очень широк. Это более 200 наследственных заболеваний почек, некоторые из которых встречаются с частотой 1 человек на 1 миллион человек и реже. Большинство нефрологов могут не встретить таких пациентов ни разу за свою жизнь.
А если встретит, наверняка, примет это за другое заболевание почек. Больные поступают к вам в запущенном состоянии?
По-разному. Почки вообще очень коварный орган. Если речь не идет об остром бактериальном воспалении, они не болят. Между тем, болезнь вершит свое разрушительное дело, вплоть до тяжелой почечной недостаточности, когда спасти ребенку жизнь может лишь гемодиализ или пересадка почки. К счастью, мы нередко успеваем предотвратить подобное развитие событий.
Какими судьбами попадают к вам пациенты?
Чаще всего – по направлению наших коллег из регионов, которые сталкиваются с затяжным и/или необычным течением болезни у ребенка, которую очень важно как можно раньше диагностировать. Как, например, Синдром Альпорта (или наследственный нефрит). Этой болезнью, которая проявляется гематурией (наличием большого количества эритроцитов в моче), чаще всего страдают мальчики. Чем раньше такой ребенок пройдет обследование, чем раньше у него будет установлен диагноз, и он начнет получать нефропротективную терапию, тем на более поздний срок удастся отодвинуть развитие у него терминальной почечной недостаточности.
Говорят, у вас в отделении такие пациенты проходят уникальное обследование…
Это правда. Одним из методов диагностики гломерулопатий у пациентов нашего отделения является биопсия почечной ткани, которую изучают тремя методами с использованием световой и электронной микроскопии, иммунофлуоресценции.
К сожалению, такой комплексный морфологический подход в комбинации с клинико-лабораторными и функциональными методами обследования в диагностике гломерулопатий используют в единичных центрах России, что не позволяет правильно диагностировать заболевание почек и нередко не определяют диагноз, который при использовании одного вида микроскопии может быть не выявлен.
А буквально два года назад у нас в Институте была создана так называемая генетическая панель, благодаря которой по анализу крови мы в течение нескольких месяцев можем с большой долей точности установить генетическую природу стероид-резистентного нефротического синдрома у детей и назначить индивидуальное (персонифицированное) эффективное лечение.
Кстати, о лечении. Наверняка при редких заболеваниях требуются и очень редкие препараты, многие из которых очень дороги. Как вы решаете эту проблему?
>Нам помогают благотворительные фонды. Такие, как фонд «АиФ.Доброе сердце», благодаря которому современное, дорогостоящее лечение получают, к примеру, наши пациенты с цистинозом – наследственным заболеванием почек, при котором дети теряют с мочой все полезное (калий, натрий, аминокислоты, белок). Они маленькие, худенькие, отстают в росте и половом развитии, у них развивается мышечная гипотония, аритмия, гипотиреоз, поражение легких, центральной нервной системы. Однако с тех пор, как 7 наших пациентов с этим редким диагнозом стали получать препарат, который предотвращает отложение кристаллов аминокислоты цистина во всех органах (в том числе – и в почечной ткани), они буквально расцвели на глазах, а многие их них стали учиться.
Немало удается и нам самим. Так, благодаря нашим усилиям, у нас теперь есть весь арсенал иммуносупрессивных средств, которые являются препаратами номер один в лечении стероид-резистентного нефротического синдрома. Не без гордости скажу, что результат применения иммуносупрессивной терапии у пациентов нашего отделения сопоставим с результатами лучших мировых центров.
Нам удается решить и эту проблему. На сей счет у нас в Институте есть этический комитет, который принимает решение о необходимости приема того или иного «редкого» препарата нашим пациентом под контролем наших специалистов, дабы избежать нежелательных эффектов. Учитывая, что у нас нередко лежат дети из Якутска, из Сахалина и других отделенных городов нашей страны, где контролировать проведение такой терапии никто не сможет, назначая любой препарат, мы всегда взвешиваем все «за» и «против».
Дети лежат у вас в отделении подолгу?
В среднем – от двух до четырех недель. За это время ребенок проходит у нас интенсивное обследование и подбор необходимого лечения.
Но, к сожалению, за одну госпитализацию провести диагностику редкого наследственного заболевания удается далеко не всегда. Да и корректировать лечение приходится постоянно. Поэтому, как правило, через 6 месяцев мы госпитализируем ребенка для оценки динамики заболевания и, при необходимости, для корректировки проводимого лечения.
Представляю, какие у вас очереди на госпитализацию! Ведь отделение не резиновое!
Больших очередей мы стараемся избегать. У нас в отделении плановая госпитализация и мы уже заранее обсуждаем с родителями наших пациентов дату следующей госпитализации и ставим ее в плановый лист.
Однако если к нам на консультацию приводят ребенка с отеками, с гипертензией, которые требуют срочного медицинского вмешательства, мы находим возможность такого ребенка госпитализировать.
А если редкий диагноз ребенку установить не удается?
Даже в этом случае мы не сдаемся. Идем методом исключения диагнозов. И нередко находим эту «иголку в стоге сена». Если нужно, связываемся с коллегами из-за рубежа. Мы сотрудничаем со многими международными организациям, лечебными учреждениями. В частности, с Левенским университетом (Бельгия), с которым мы заключили двухлетний контракт на совместные исследования и ведение протоколов лечения редких наследственных заболеваний почек у детей.
Каждый больной – это поиск, это головоломка, решить которую – значит спасти жизнь и существенно улучшить ее качество.
Морфология и наследственность синдрома Альпорта
Морфология и наследственность синдрома Альпорта
Почечные изменения при синдроме Альпорта демонстрируют комбинированные черты хронического гломерулонефрита, пиелонефрита и интерстициального нефрита, но с недостаточно полной характеристикой каждого из этих заболеваний.
Почки маленьких размеров имеют мелкозернистое и палисадное строение. На срезе поверхности коры могут быть обнаружены желтые линейные прожилки. Первичные изменения встречаются в соединительнотканной мембране клубочков.
Наиболее ранним поражением являются выраженная пролиферация эпителия в клубочках, интерстициальный фиброз или локальное расширение с атрофией канальцев, появляющиеся приблизительно одновременно. В большинстве случаев наблюдаются заполненные липидами пенистые клетки, не связанные с эпителиальными клетками канальцев. Эти пенистые клетки могут заполнять интерстициальную ткань, особенно в нижних отделах коры, располагаясь в виде рядов или гнезд. В большинстве случаев в интерстициальной ткани находят плазматические клетки, лимфоциты и отложения кальция. Капальцевыс изменения включают атрофию эпителия и расширение некоторых канальцев. Иногда наблюдается хронический интерстициальный нефрит, при котором изначально мало клубочков (White et al., Krickstein et al.).
Необходимо подчеркнуть, что, в то время как все эти изменения обусловлены синдромом Альпорта, они не являются специфичными для него. Исследование ультраструктуры установили характерное расслоение соединительнотканной мембраны клубочков па большое число тонких пластинок в результате накопления между слоями маленьких плотных частичек (Churg a. Sherman, Scherman et al.).
Патогенез. Патогенез синдрома Альпорта неизвестен. Quick с соавт., Arnold и Weidaver отметили сходство улитки и почек по некоторым пунктам (баланс жидкости и электролитов, общие ототоксические и нефротоксические вещества). Они продемонстрировали иммунохимические и иммуногистохимическис доказательства сходства антигенов в почках и в улитке.
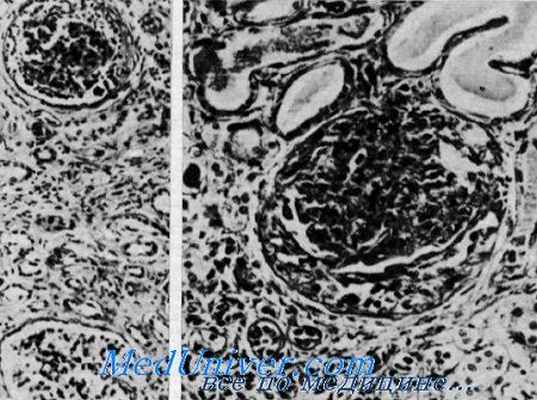
Нефрит и нейросенсорная глухота (синдром Альпорта). Сверху клубочек с утолщенной основной мембраной и слипшейся капсулой в противоположность почти нормальному клубочку внизу. Видны редкие клубочки, имеющие полулупиую форму, расширенные канальцы, содержащие белковые цилиндры. Утолщенная основная мембрана, окружающая атрофичные канальцы в области фиброза (из H. I. Krickstein et al.).
Наследственность. В некоторых исследованиях было высказано предположение, что гетерозиготные матери передают аномальный ген примерно 65% своих детей. Среди потомства больных мужчин синдром Альпорта отмечается у 75% их дочерей и только у 45% их сыновей. Cohen с соавт., Shaw и Glover, MacNeill и Shaw предположили, что синдром определяется одним аутосомным геном, который регулярно наследуется у женщин (имея сродство скорее к овоцитам, чем к полярным тельцам) и наследуется преимущественно с Х-хромосомой при сперматогенезе.
Preus и Fraser предположили, что неблагоприятная внутриутробная среда больной матери может быть причиной повышенной ненетрантности гена у ее сыновей. Мы согласны с Мауо в том, что синдром Альпорта, вероятно, генетически гетерогенен.
Диагноз. Синдром Альпорта должен быть отграничен от всех других случаев гематурии и иротеинурии в детском возрасте. Гематурией сопровождаются также гломерулопефрит и гломерулит. Наследственный нефрит без глухоты, описанный Dockhorn, является, по-видимому, другим заболеванием.
Лечение. Глухота обычно уменьшается при пользовании слуховыми аппаратами. Терапия должна быть направлена на коррекцию биохимических нарушений, сопровождающих почечную недостаточность.
Необходимо обсудить вопрос о пересадке почек. У одного больного в результате этой операции мы наблюдали улучшение слуха.
Прогноз. Прогноз варьирует. У некоторых больных заболевание может протекать легко и практически не оказывать влияния на их жизнь, в то время как у других больных может развиться тяжелая почечная недостаточность, от которой они погибают в молодом возрасте.
Выводы. Характеристика этого синдрома включает: 1) аутосомнодоминантное наследование с более тяжелым поражением мужчин; 2) прогрессирующий нефрит с уремией; 3) патологию хрусталика, включающую лентиконус, сферофакию или катаракты, примерно в 10% случаев; 4) начинающуюся в нервом или во втором десятилетии жизни прогрессирующую пейросенсорную глухоту с варьирующей экспрессивностью.
Синдром Альпорта
Синдром Альпорта - генетически гетерогенное заболевание, характеризующееся нефритическим синдромом (т.е. гематурией, протеинурией, гипертонией, в конечном итоге почечной недостаточностью), часто с нейросенсорной глухотой и, реже, офтальмологическими симптомами. В основе заболевания лежит генная мутация, повреждающая коллаген IV типа. Диагностика производится на основании данных анамнеза, включая семейный анамнез, результатов анализа мочи и биопсии (почки или кожи). Лечение такое же, как и при хронической болезни почек, иногда включает пересадку почки Трансплантация почки Трансплантация почек является наиболее распространенным видом трансплантации цельных органов. (См. также Обзор трансплантации (Overview of Transplantation)). Основное показание для трансплантации. Прочитайте дополнительные сведения .
Синдром Альпорта – это нефритический синдром, вызванный мутацией гена COL4A3, COL4A4, COL4A5, (кодирующего альфа-5 цепь коллагена IV типа), что приводит к формированию измененных коллагеновых волокон IV типа. Механизм развития гломерулярной болезни в результате изменений структуры коллагена неизвестен, но предполагают, что в основе лежит нарушение структуры и функции; у членов большинства семей развивается утолщение и истончение гломерулярных и канальцевых базальных мембран с образованием многослойной плотной пластинки с очаговым или местным распределением (пример переплетения "плетенка"). В результате происходит рубцевание гломерулярной ткани и интерстициальный фиброз.
Болезнь обычно наследуется по Х-сцепленному механизму, хотя существуют аутосомно-рецессивные и, редко, аутосомно-доминантные варианты. Случаи наследования по Х-хромосоме могут быть клинически классифицированы как:
Ювенильная форма: почечная недостаточность развивается между 20 и 30 годами
Зрелая (взрослая) форма: почечная недостаточность развивается у людей > 30 лет
Клинические проявления
Классическое Х-хромосомное заболевание у мужчин и аутосомно-рецессивное заболевание клинически сходны. У пациентов развиваются почечные симптомы, характерные для острого нефритического синдрома (например, микрогематурия, гипертензия, а впоследствии макрогематурия с протеинурией), а прогрессирование заболевания до почечной недостаточности происходит в возрасте 20–30 лет (ювенильные формы).
Нейросенсорная тугоухость встречается часто, при этом характерно нарушение восприятия более высоких частот; она может оставаться недиагностированной в раннем детстве.
Реже, чем тугоухость, наблюдаются офтальмологические патологии: катаракта (чаще всего), передний лентиконус (простая протрузия конуса к переднему полюсу хрусталика из-за утолщения капсулы хрусталика), сферофакия (сферическая деформация хрусталика, предрасполагающая к подвывиху хрусталика), нистагм, пигментный ретинит, слепота.
Х-хромосомное заболевание встречается у гетерозиготных женщин, которые, потому что они имеют одну нормальную Х-хромосому, как правило, имеет менее тяжелые, более медленно прогрессирующие симптомы, чем у мужчин.
У некоторых мужчин с Х-хромосомным заболеванием почечная недостаточность развиваются после 30 лет с потерей слуха, которая происходит с опозданием или в легкой форме, а аутосомно-доминантное заболевание, как правило, не приводит к почечной недостаточности до возраста ≥ 45 лет (взрослые формы).
У больных с Х-хромосомным синдромом Альпорта, сенсоневральная потеря слуха, обычно, проявляется в детсве, в то время, как болезнь почек часто не проявляется до взрослого возраста.
Морфология и наследственность синдрома Альпорта
Впервые описан в 1927 году Альпортом как сочетание наследственного нефрита с глухотой. Описано 6 вариантов синдрома Альпорта, которые наследуются по сцепленному с Х-хромосомой доминантному или рецессивному типу (ген картирован на длинном плече Х-хромосомы q22, ответственен за синтез 4А5 коллагена), или аутосомно-доминантному, или аутосомно-рецессивному типу (ген картирован на 2-й хромосоме).
В основе болезни лежит нарушение образования трехспиральной структуры коллагена 4 типа, в том числе базальных мембран клубочков, аналогичных структур уха и глаза. При морфологических исследованиях выявляют фокально-сегментарный гломерулосклероз, мембранозно-пролиферативные, мезангио-пролиферативные изменения, атрофию и дистрофию канальцев, интерстициальный фиброз. Во внутреннем ухе находят потерю нейронов и волосяных клеток, атрофию спиральных связок, поражения 8 пары черепно-мозговых нервов, Кортиева органа. Со стороны глаз с разной частотой обнаруживают снижение остроты зрения, передний лентиконус, пятна на сетчатке, кератоконус, катаракту. При биохимических исследованиях мочи характерно преобладание дерматансульфата, глюкозилгалактозилоксолизина при уменьшении экскреции оксипролина. У части больных обнаружены также снижение уровня в крови IgA, T- и B-лимфоцитов, фагоцитарной активности.
Отмечается выраженный клинический полиморфизм. Наиболее характерным является гематурия, протеинурия, периодическая бактериурия, снижение слуха. Первые признаки синдрома Альпорта появляются обычно в возрасте 5-10 лет. Выраженность мочевого синдрома поначалу минимальна, нет нарушений функции почек. В дальнейшем постепенно появляются и неуклонно нарастают явления ХПН (более быстро и тяжело у лиц мужского пола). Снижение слуха может развиться еще до появления почечной патологии. В начале это нейросенсорное снижение слуха высоких тонов, далее -- низких, переходящее из звукопроводящей в звуковоспринимающую тугоухость. Могут быть также миастения, потеря памяти и интеллекта, тромбоцитопения. В семьях больных описаны лица с изолированной тугоухостью, доброкачественной гематурией.
Важно составление родословной и обнаружение лиц как с сочетанием нефрита и глухоты, так и с доброкачественной гематурией, тухоухостью, ХПН. Диагноз подтверждают при биопсии почек, обнаружении в моче D/L-3-гидроксипролина, глюкозилгалактозилоксилизина. У части больных выявляют сужение прилоханочного отдела мочеточника, удвоение, незавершенный поворот почек.
Назначение глюкокортикоидов и цитостатиков неэффективно и лишь ухудшает прогноз. Диета высококалорийная в соответствии с возрастом и функциональным состоянием почек. Показано раннее выявление и активное лечение как мочевой инфекции, так и хронических очагов инфекции, последовательные курсы "трофических" препаратов -- АТФ, кокарбоксилазы, пиридоксина, ксидифона, витаминов А, Е, В5, В15 etc., фитотерапия. Некоторые авторы описывают положительный эффект от применения делагила в дозе 5-10 мг/кг в сутки в течение 6-12 мес, левамизола (2,5 мг/кг в сутки 2-3 раза в неделю в течение 4-6 нед). При начальных признаках ХПН показана трансплантация почки, хотя у 1-5% после трансплантации может развиться нефрит с антителами к базальной мембране клубочков.
Морфологическая характеристика синдрома Альпорта
В данной статье отражены классические морфологические проявления синдрома Альпорта на примере данных 18 нефробиопсий детей от 4 до 18 лет с данной патологией.
Ключевые слова: гломерулонефрит, нефробиопсия, синдром Альпорта.
СиндромАльпорта (СА)— этиологически гетерогенное наследственное заболевание моногенной природы. Причина заболевания лежит в мутации одного из генов: COL4A5, COL4A4, COL4A3. При классическом варианте СА мутация происходит в гене COL4A5, расположенном на длинном плече Х-хромосомы (Хq22.2), который кодирует 5-цепь коллагена IV типа (НГ).
Распространенность СА составляет 1:5000. Он лежит в основе развития 1 % всех случаев хронической почечной недостаточности (ХПН) в Европе. 2,3 % случаев трансплантации почки проводится больным с СА [1].
Классические изменения почечной паренхимы при синдроме Альпорта представляют сочетание морфологических черт хронического гломерулонефрита, пиелонефрита и интерстициального нефрита, но с неполной картиной каждой из патологий. Почки уменьшены в размерах, их структура мелкозернистая или палисадная. На срезе коркового вещества часто имеются желтые линейные прожилки. Первичные изменения наблюдаются в гломерулярной мембране. Наиболее ранним поражением являются выраженная пролиферация эпителия в клубочках, интерстициальный фиброз или локальное расширение с атрофией канальцев, появляющиеся приблизительно одновременно. В большинстве случаев наблюдаются заполненные липидами пенистые клетки, не связанные с эпителиальными клетками канальцев. Эти пенистые клетки могут заполнять интерстициальную ткань, особенно в нижних отделах коры, располагаясь в виде рядов или гнезд. В большинстве случаев в интерстициальной ткани находят плазматические клетки, лимфоциты и отложения кальция. Канальцевые изменения включают атрофию эпителия и расширение некоторых канальцев. Иногда наблюдается хронический интерстициальный нефрит, при котором изначально мало клубочков.
При анализе нефробиопсий 18 пациентов на базе патологоанатомического бюро 3-ей городской детской клинической больницы г. Минска в большинстве случаев (83 %) обнаружен диффузный или фокальный, глобальный или сегментарный мезангиопролиферативный гломерулонефрит. У 17 % обследованных выявленные изменения расценены как фокально-сегментарный гломерулосклероз. Тубулоинтерстициальные изменения включали обнаруженные во всех случаях пенистые клетки, одиночные и сформировавшиеся в виде кластеров в интерстиции и канальцах, 39 % случаях — очаговый межуточный склероз с атрофией канальцев, в 28 % случаев — очаговое межуточное воспаление разной степени выраженности. При иммуногистохимии в большинстве случаев (61 %) экспрессия иммуноглобулинов A, G, M и С3 С1q фракций комплемента отсутствовала, в 33 % случаев выявлена экспрессия иммуноглобулина M, в 11 % случаев — экспрессия иммуноглобулина M и С3 С1q фракций комплемента. У 8 пациентов, которым проведено иммуногистохимическое исследование с применением антител к α3 и α5 субъединицам коллагена IV типа, выявлена гетерогенность иммуногистохимического окрашивания — от полного отсутствия до сохранения обеих субъединиц.
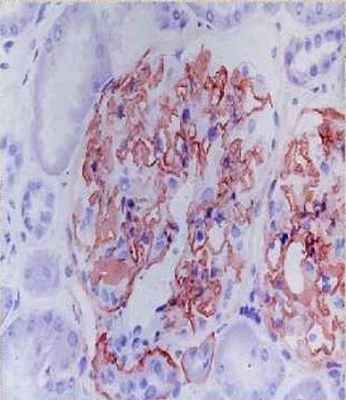
Рис. 1. Иммуногистохимическое исследование с применением антител к α3 (контроль)
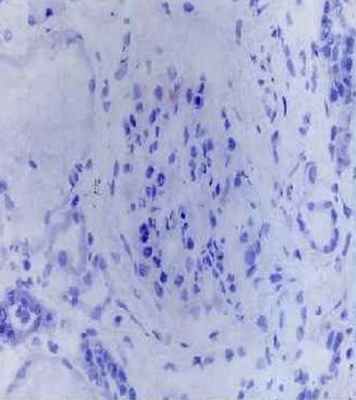
Рис. 2. Иммуногистохимическое исследование с применением антител к α3 (СА)
Таким образом, синдром Альпорта в большинстве случаев морфологически проявляется мезангиопролиферативным гломерулонефритом с наличием пенистых клеток и отсутствием экспрессии иммуноглобулинов A, G, M и С3 С1q фракций комплемента.
- Atkin C. L., Gregory V. S., Border W. A. Alport syndrome. In: R. W. Schrier, C. M. Cottschalk (eds.) Diseases of the Kidney. 4th ed.Boston: Little 1989; 233.
Основные термины (генерируются автоматически): большинство случаев, фракция комплемента, изменение, интерстициальная ткань, клетка, экспрессия иммуноглобулина.
Читайте также: