Диагностика и лечение аутосомно-рецессивного поликистоза почек
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Диагностика и лечение аутосомно-рецессивного поликистоза почек
Диагноз аутосомно-рецессивного поликистоза почек основывается на клинической картине, данных лучевой диагностики и биопсии почек и печени, а также генодиагностики. При длительном течении заболевания появляется еще один диагностический признак — кистозное расширение внутрипеченочных желчных протоков (болезнь Кароли).
Лучевая диагностика и биопсия аутосомно-рецессивного поликистоза почек
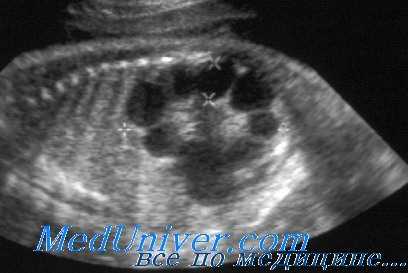
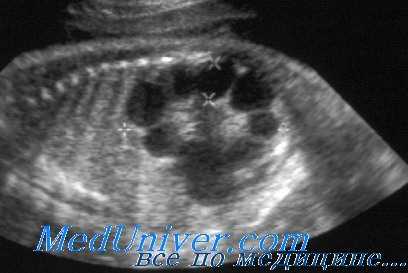
При УЗИ почки резко увеличены, но форма их сохранена. Паренхима имеет повышенную эхогенность, крупнозернистая, с наличием мелких эхонегативных зон («соль с перцем»). На более поздней стадии помимо веретенообразных могут появиться кисты сферической формы. УЗИ печени выявляет повышенную эхогенность, а позднее — хорошо различимые кисты желчных протоков.
Диагностике помогают данные КТ и МРТ почек и печени и экскреторной урографии.
При МРТ собирательные трубочки расширены в виде веретенообразных, радиально расположенных кист, но решающего значения при постановке диагноза эти данные не имеют. В кисты превращены от 10 до 90% собирательных трубочек — в зависимости от тяжести и возраста начала заболевания. При биопсии печени находят разрастание желчных протоков, портальный и перипортальный фиброз.
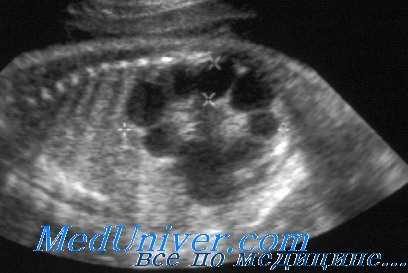
УЗИ при поликистозе почек
Медико-генетическое консультирование и генодиагностика аутосомно-рецессивного поликистоза почек
Ген, ответственный за аутосомно-рецессивный поликистоз почек, находится на 6-й хромосоме (сегмент 6р21.1—р12). Локусной генетической гетерогенности не выявлено даже для поликистоза с ранним началом (в перинатальном периоде). Так как тип наследования аутосомно-рецессивный, риск заболевания для родных братьев и сестер больного составляет 25%.
В пренатальном периоде диагноз можно заподозрить при маловодий, а затем подтвердить или опровергнуть его с помощью УЗИ. Однако на ранних сроках беременности УЗИ недостаточно информативно, поскольку почки плода могут начать увеличиваться лишь с 24—28 нед внутриутробного развития. Иными словами, отрицательный результат УЗИ, проведенного на ранних сроках беременности, не исключает аутосомно-рецессивный поликистоз почек у плода.
Чтобы отличить аутосомно-рецессивный поликистоз от аутосомно-доминантного с ранним началом, обязательно проводят УЗИ почек родителей больного ребенка. Поскольку сам ген, ответственный за аутосомно-рецессивный поликистоз, не идентифицирован, а известно лишь его положение на 6-й хромосоме, то при наличии хотя бы одного больного в семье для генодиагностики можно воспользоваться анализом гаплотипов.
Дифференциальная диагностика аутосомно-рецессивного поликистоза почек
Дифференциальная диагностика аутосомно-рецессивного поликистоза почек требует исключения других заболеваний, которые проявляются в грудном возрасте и сопровождаются кистами почек. При мультикистозе почки уменьшены, форма их нарушена, характерны пороки развития мочевых путей. Чтобы в столь раннем возрасте проявился аутосомно-доминантный поликистоз, в гене PKD1 должны произойти крупные делении нуклеотидов.
Синдромы Лоренса—Муна и Барде—Бидля легко отличить от аутосомно-рецессивного поликистоза почек по таким дополнительным признакам, как пигментная дегенерация сетчатки, полидактилия, ожирение, умственная отсталость и гипогонадизм. Для синдрома Меккеля—Грубера помимо агенезии или дисгенезии почек характерен широкий спектр внепочечных проявлений, в том числе энцефалоцеле в затылочной области, расщелина неба и пороки развития глаз.
Двустороннюю нефробластому (опухоль Вильмса) отличают от аутосомно-рецессивного поликистоза с помощью лучевой диагностики — опухолевая ткань резко отличается от нормальной паренхимы почек.
Лечение аутосомно-рецессивного поликистоза почек
Иногда проводят трансплантацию печени — либо одновременно с трансплантацией почки, либо, если показания сохраняются, позднее.
Прогноз аутосомно-рецессивного поликистоза почек
До 1 года доживают 94% мальчиков и 82% девочек, родившихся с аутосомно-рецессивным поликистозом почек. Интересно, что у родных братьев и сестер почечная недостаточность при аутосомно-рецессивном поликистозе почек развивается практически в одном и том же возрасте.
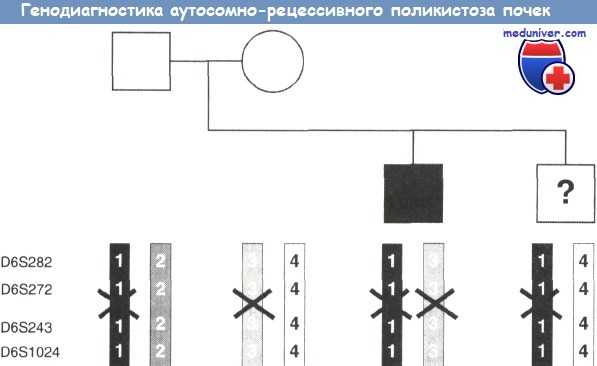
Ген, ответственный за АРПП, расположен на 6-й хромосоме. Но так как точная локализация гена на хромосоме неизвестна, то с помощью ПЦР выявляют группы полиморфных участков ДНК, расположенных рядом с предполагаемым геном АРПП. Такая группа полиморфных участков на каждой из гомологичных хромосом называется гаплотипом. Анализ гаплотипов позволяет установить, из какой гаметы — материнской или отцовской — происходит мутация, и проследить ее передачу потомству, даже если точное положение гена на хромосоме не установлено. В этой семье (родословная изображена в верхней части рисунка) родился мальчик с АРПП. Цель исследования — определить, будет ли болен АРПП и второй ребенок. На рисунке показаны четыре исследованных полиморфных участка ДНК, полученных от четырех членов семьи. Все гаплотипы пронумерованы, а материнские и отцовские хромосомы обозначены разным цветом. Ген АРПП расположен между участками D6S272 и D6S243 хромосомы. Больной мальчик получил по одному гаплотипу, содержащему мутантный ген (указаны крестиками), от матери и от отца. Второй же ребенок в семье получил мутантный ген лишь с отцовской гаметой, в то время как со стороны матери ген не поврежден. Таким образом, этот ребенок будет гетерозиготным носителем гена АРПП и болезнь у него не проявится.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
- Вернуться в раздел "нефрология"
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Аутосомно-рецессивный поликистоз почек у детей. Диагностика и лечение
Многие врожденные аномалии мочевых путей могут сопровождаться макро- или микрогематурией. Внезапная макрогематурия после небольшой травмы поясничной области характерна для обструкции лоханочно-мочеточникового сегмента или поликистоза почек.
Аутосомно-рецессивный поликистоз почек - это заболевание, называемое также поликистозной болезнью младенцев, встречается с частотой 1:10000-1:40 000. Ген, ответственный за аутосом-но-рецессивный поликистоз почек, пока не найден, но исследования по генетическому сцеплению позволяют локализовать его на коротком плече хромосомы 6.
Патоморфология аутосомно-рецессивного поликистоза почек. Обе почки резко увеличены, содержат много кист в корковом и мозговом веществе. Под микроскопом видны многочисленные кисты, распространяющиеся от мозгового вещества в корковое и расположенные преимущественно в собирательных трубочках и протоках. У плодов наблюдаются преходящие кисты и в проксимальных канальцах. Прогрессирующий фиброз интерстициальной ткани и атрофия канальцев в конце концов приводят к почечной недостаточности.
Поражение печени характеризуется пролиферацией и эктазией желчных протоков, а также фиброзом, неотличимым от врожденного фиброза печени или болезни Кароли (расширение внутрипеченочных протоков с холелитиазом, рецидивирующим холангитом и желтухой).
Клинические проявления аутосомно-рецессивного поликистоза почек. В типичных случаях у новорожденных или грудных детей обнаруживаются объемные образования в боковых отделах живота. Аутосомно-рецессивный поликистоз почек может сопровождаться маловодием, гипоплазией легких, респираторным дистресс-синдромом и спонтанным пневмотораксом в неонатальном периоде. Возможны аномалии лицевого скелета (лицо Поттера) и другие последствия уменьшения объема околоплодных вод, включая низко посаженные уши, микрогнатию, уплощенный нос, контрактуру конечностей и дефицит роста.
В первые несколько недель жизни обычно развивается высокая артериальная гипертония. Объем мочи, как правило, не снижен, хотя иногда отмечается олигурия и острая почечная недостаточность. В неонатальном периоде обычно имеет место преходящая гипонатриемия (часто на фоне острой почечной недостаточности), которую можно снять диуретиками. У 20-30 % больных функция почек остается нормальной. В редких случаях аутосомно-рецесивный поликистоз почек проявляется почечной недостаточностью, артериальной гипертонией или гепатоспленомегалией (вследствие фиброза печени) в более позднем возрасте.
Лучевые или лабораторные методы исследования определяют поражение печени примерно у 45 % новорожденных с этим заболеванием. Однако у больных повышен риск развития 1) восходящего холангита, варикозного расширения вен и гиперспленизма из-за портальной гипертензии и 2) прогрессирующей дисфункции печени, которая может привести к явной печеночной недостаточности и циррозу.
У части старших детей с аутосомно-рецессивным поликистозом почек заболевание проявляется именно гепатоспленомегалией, а поражением почек обнаруживается случайно при рентгенографии брюшной полости.
Диагностика аутосомно-рецессивного поликистоза почек. Пальпируемое двустороннее объемное образования в боковых отделах живота у грудного ребенка с гипоплазией легких, маловодием и артериальной гипертонией в отсутствие поликистоза почек у родителей позволяет диагностировать болезнь. При УЗИ почки обычно резко увеличены и равномерно повышена их эхогенность со стертостью границы между корковым и мозговым веществом. Диагноз подтверждают также клинические и лабораторные признаки фиброза печени, патологические изменения желчных протоков в биоптате печени, наличие поликистоза почек у сиблингов или близкое родство родителей. Аутосомно-рецессивный поликистоз почек следует отличать от увеличения почек при поликистозной дисплазии, гидронефроза, опухоли Вильмса и двустороннем тромбозе почечных вен. В семьях хотя бы с одним больным ребенком возможна пренатальная диагностика с помощью анализа генетического сцепления и использования информативных маркеров.
Лечение аутосомно-рецессивного поликистоза почек симптоматическое. Гипоплазия легких и гиповентиляция в неонатальном периоде часто требуют ИВЛ. Необходима нормализация АД и водно-электролитного баланса, устранение клинических проявлений почечной недостаточности. При тяжелой дыхательной недостаточности и в случаях, когда увеличенные почки препятствуют усвоению пищи, может потребоваться нефрэктомия с последующим диализом.
Прогноз аутосомно-рецессивного поликистоза почек. Около 30 % больных погибает в неонатальном периоде от осложнений гипоплазии легких. Однако, если дети не погибают в первой год жизни, то современные методы лечения дыхательной и почечной недостаточности у новорожденных увеличивают 10-летнюю выживаемость до 80 % и более. Терминальная стадия ХПН (обычно в первые 10 лет жизни) наблюдается более чем в 50 % случаев. Стандартные методы лечения детей — диализ и трансплантация почки. Последующие заболеваемость и смертность связаны с осложнениями ХПН и поражением печени.
Поликистоз почек ( Поликистозная болезнь почек )
Поликистоз почек – это врожденная двусторонняя кистозная трансформация почечной паренхимы, приводящая к прогрессирующему снижению функции почек. Патология может проявляться артериальной гипертензией, болевым синдромом в поясничной области и животе, гематурией, дизурией, развитием инфекции и камней в почках, почечной недостаточностью. Диагностика заболевания включает изучение семейного анамнеза, лабораторные исследования, УЗИ почек, урографию, ангиографию, МРТ и КТ. Лечение при поликистозе почек консервативное, симптоматическое.
МКБ-10
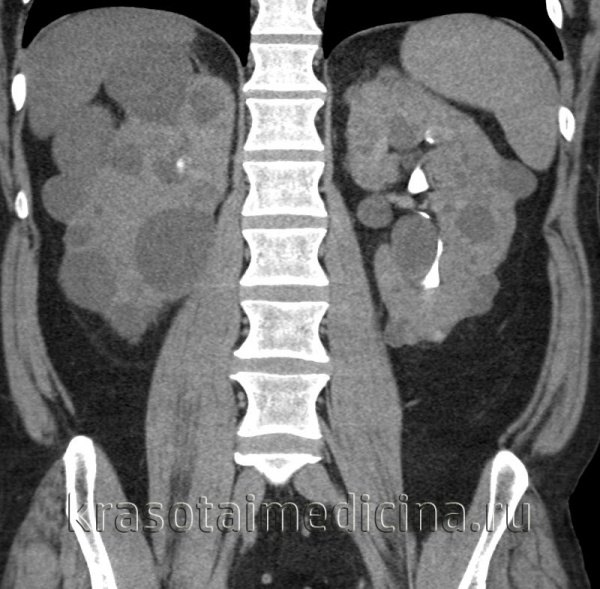
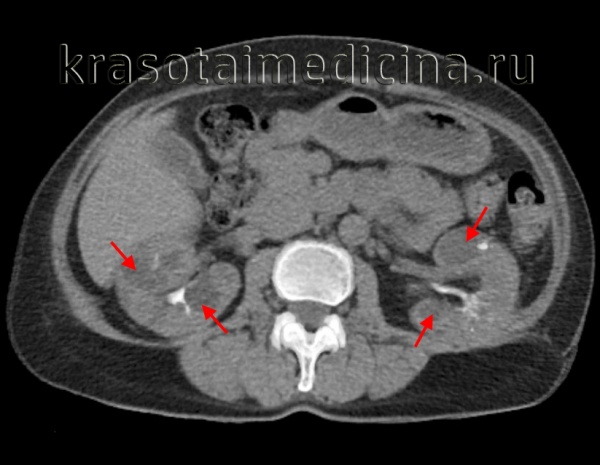
Общие сведения
Поликистозная болезнь (поликистоз почек) – порок эмбрионального развития почечных канальцев, характеризующийся образованием множественных мелких кист в паренхиме почек. Поликистоз почек всегда двусторонний. Кисты могут иметь размер от спичечной головки до крупной вишни и больше, содержать светлый либо шоколадного цвета желеобразный секрет.
Увеличение кист со временем приводит к сокращению объема функционирующей паренхимы и развитию почечной недостаточности. Как правило, поликистоз не ограничивается только поражением почек; к внепочечным формам поликистоза относится формирование кист в поджелудочной железе, селезенке, печени, семенных пузырьках, легких и других органах.
Причины
Поликистоз почек в большинстве случаев является патологией, наследуемой по аутосомно-доминантному типу. Развитие аутосомно-доминантного поликистоза может произойти, если аналогичное заболевание имеется у одного из родителей; эта форма наследования определяет 85-90% случаев болезни. Признаки заболевания в этом случае чаще развиваются к 30-40 годам, реже в детском возрасте.
Для развития аутосомно-рецессивного поликистоза почек необходима передача мутантных генов, обусловливающих дефект, от обоих родителей. Это менее распространенная форма, которая обнаруживается у новорожденных. При отсутствии семейной истории предполагается возникновение новой мутации в половой клетке одного из родителей.
Патогенез
В патогенезе поликистоза почек ведущая роль принадлежит нарушению слияния секреторных и экскреторных структур нефрона на стадии развития вторичной почки. Это затрудняет выделение первичной мочи, увеличивает давление в почечных канальцах, что приводит к деформации их просвета и образованию кистозно расширенных полостей. Отмечается значительное увеличение размеров и массы почек, которые макроскопически имеют бугристую неровную поверхность из-за множественных выступающих кист.
Стенки кист представлены соединительной тканью, полость выстлана плоским или кубическим эпителием, внутри содержится жидкость желтоватого или коричневого цвета, близкая по составу к моче. Между отдельными кистами имеются участки паренхимы, которые из-за давления кист могут подвергаться дистрофическим изменениям, атрофии или ишемии. Чашечки и лоханки значительно деформированы и увеличены.
Классификация
Симптомы поликистоза почек
У новорожденных патология обычно протекает крайне неблагоприятно и достаточно рано заканчивается гибелью ребенка от уремии. У взрослых поликистоз почек развивается медленно, проходя компенсированную, субкомпенсированную и декомпенсированную стадии.
На стадии компенсации проявления длительно отсутствуют. Со временем появляется чувство давления в пояснице, неопределенные боли в животе, дизурия, обусловленные растяжением почек. Отмечается утомляемость, головная боль, иногда – гематурия неясного генеза. Функция почек в компенсированной стадии остается не нарушенной.
В стадии субкомпенсации нарастают признаки почечной недостаточности, проявляющиеся тошнотой, сухостью во рту, жаждой, приступами мигрени, стойкой и высокой артериальной гипертензией. Нарушения функции почек характеризуются полиурией с изостенурией, эритроцитурией, цилинрурией, при возникновении пиелонефрита – лейкоцитурией. В случае нагноения кист присоединяется лихорадка, интоксикация, ознобы; при камнях в почках развиваются приступы почечной колики.
В декомпенсированной стадии болезни возникает хроническая уремия. Прогрессированию поликистоза почек может способствовать артериальная гипертензия, травмы, хирургические вмешательства, беременность, кровотечения. Присоединение вторичной инфекции (гриппа, ОРВИ, пневмонии и др.) может вызвать резкое ухудшение состояния вплоть до гибели пациента; при нагноении кист нередко развивается уросепсис.
Осложнения
Стойкое повышение кровяного давления со временем может осложняться гипертрофией левого желудочка, пролапсом митрального клапана и сердечной недостаточностью, аневризмой сосудов мозга и геморрагическим инсультом. Поликистоз почек может вызывать развитие поздних токсикозов беременности – преэклампсии и эклампсии. В группе особого риска – женщины, страдавшие гипертонией до наступления беременности. У пациентов с этой почечной патологией повышена вероятность образования кист печени, дивертикулярной болезни толстого кишечника.
Диагностика
Данные анамнеза в ряде случаев позволяют выявить семейные случаи поликистоза почек у родственников одной линии. Пальпировать увеличенные и кистозно измененные почки удается не всегда, в связи с чем решающее значение в диагностике отводится инструментальным методикам:
Посредством УЗИ в увеличенных почках определяются множественные кисты. В неясных случаях прибегают к ретроградной пиелографии, почечной ангиографии, которые также позволяют обнаружить кистозное перерождение почек. Для выяснения степени компенсации функции почек проводят исследования мочи (общий анализ, пробу Зимницкого и Реберга), биохимическое исследование крови. При развитии пиелонефрита моча подвергается бактериологическому посеву. С целью установления семейных форм поликистоза почек показано генетическое исследование.
Поликистоз почек необходимо дифференцировать от хронического гломерулонефрита и хронического пиелонефрита, опухолей почки. При подозрении на аневризму сосудов головного мозга выполняется ангиография сосудов головного мозга, УЗДГ, магнитно-резонансная ангиография.
Лечение поликистоза почек
Проводится симптоматическая терапия. К общим рекомендациям относится исключение чрезмерных и длительных нагрузок, профилактика хронических инфекций (кариеса, ОРВИ, тонзиллита, синуситов и др.), соблюдение высококалорийной, богатой витаминами диеты с ограничением белка и поваренной соли. При развитии пиелонефрита назначается курсовое лечение антибиотиками и уроантисептиками. В случае макрогематурии осуществляется гемостатическая терапия; при снижении диуреза показан прием диуретиков; при артериальной гипертонии – гипотензивных средств.
В компенсированной стадии поликистоза почек может выполняться вскрытие и опорожнение крупных кист. Это приводит к уменьшению размеров почек, улучшению их кровообращения и функций. В терминальных стадиях, при почечной недостаточности ставится вопрос о хроническом гемодиализе и трансплантации почки.
Прогноз и профилактика
Своевременная коррекция артериальной гипертензии и устранение инфекций мочевых путей позволяют замедлить прогрессирование поликистоза почек. Тем не менее, у большинства больных с поликистозом почек в различные сроки от выявления болезни развивается почечная недостаточность. При семейных формах поликистоза почек необходима консультация врача-генетика для определения рисков рождения ребенка с подобной почечной аномалией. При установленном диагнозе требуется постоянное наблюдение пациента врачом-нефрологом и урологом.
Поликистоз почек аутосомно-доминантный (АДПКП): причины, диагностика, лечение
Этиология и встречаемость поликистоза почек. Аутосомно-доминантный поликистоз почек (АДПКП) (MIM №173900) — генетически разнородное заболевание. Приблизительно у 85% пациентов поликистоз I типа вызван мутациями в гене PKD1; большинство остальных имеют поликистоз II типа вследствие мутаций в гене PKD2. Несколько семей не показывают сцепления с этим локусами, что позволяет предполагать наличие, по крайней мере, еще одного дополнительного, пока неопознанного локуса.
Патогенез аутосомно-доминантного поликистоза почек (АДПКП)
Ген PKD1 кодирует полицистин-1, трансмембранный рецептороподобный белок неизвестной функции. Ген PKD2 кодирует полицистин-2, мембранный белок, гомологичный натриевому и кальциевому а1 каналам. Полицистин-1 и полицистин-2 взаимодействуют как часть гетеромногомерного комплекса.
Образование кист при аутосомно-доминантном поликистозе почек (АДПКП), как оказалось, соответствует «двухударному» механизму, наблюдающемуся при мутациях генов-супрессоров опухолевого роста и новообразованиях; т.е. для формирования кист должны утратить функцию оба аллеля гена PKD1 или PKD2. Механизм функциональной причины образования кисты при утрате функций полицистинов до конца не определен, но включает неправильное расположение белков на поверхности клетки, в норме ограниченное базолатеральной или эпителиальной поверхностью формирующихся тубулярных клеток почки.
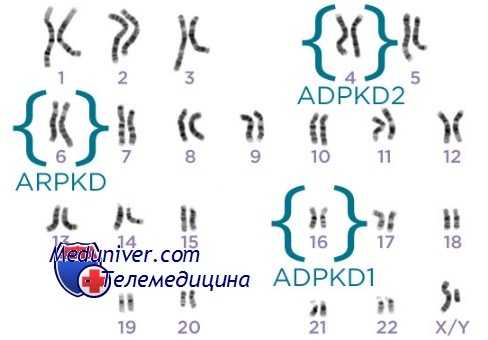
Фенотип и развитие аутосомно-доминантного поликистоза почек (АДПКП)
Аутосомно-доминантный поликистоз почек (АДПКП) может обнаруживаться в любом возрасте, но симптомы или признаки чаще появляются на третьем-четвертом десятилетиях жизни. У пациентов появляются инфекции мочевых путей, гематурия, нарушения уродинамики (сгустки или камни почек), ноктурия, кровоизлияния в кисты или жалобы на боли в боку, вызванные увеличением размеров расширенных почек. Артериальная гипертензия бывает у 20-30% детей и почти у 75% взрослых с аутосомно-доминантным поликистозом почек. Гипертония — вторичный эффект внутрипочечной ишемии и активизации системы ренин-ангиотензин. Почти половина больных к 60 годам жизни имеет почечную недостаточность. Гипертония, повторные инфекции мочевых путей, мужской пол и возраст начала клинических проявлений — наиболее важные факторы, указывающие на раннее развитие почечной недостаточности. Приблизительно 43% пациентов с началом болезни вскоре после рождения умирают от почечной недостаточности в течение первого года жизни; у остальных к 30 годам жизни формируется терминальная почечная недостаточность, гипертония или и то, и другое вместе.
Аутосомно-доминантный поликистоз почек (АДПКП) демонстрирует как межсемейную, так и внутрисемейную изменчивость по возрасту начала и тяжести. Часть междусемейной изменчивости вторична относительно локусной гетерогенности, поскольку пациенты с поликистозом II типа имеют более мягкие проявления болезни, чем пациенты с I типом болезни. Внутрисемейная изменчивость, как оказалось, вызвана совместным влиянием среды и генетического фона, поскольку изменчивость более выражена между поколениями, чем среди сибсов.
Кроме кист почек, у пациентов с аутосомно-доминантным поликистозом почек образуются кисты в печени, поджелудочной железе, яичниках и селезенке, а также внутричерепные аневризмы, пролапс митрального клапана и дивертикулы толстой кишки. Кисты печени часто встречаются как при АДПКП-1, так и при АДПКП-2, тогда как кисты поджелудочной железы обычно наблюдают при АДПКП-1. Внутричерепные мешотчатые аневризмы выявляют у 5-10% больных АДПКП; тем не менее риск развития аневризм неодинаков для всех пациентов, поскольку они имеют семейное накопление. Больные аутосомно-доминантным поликистозом почек имеют повышенный риск недостаточности аортального и трехстворчатого клапанов, а пролапс митрального клапана выявляют у 25% больных. Дивертикулы толстого кишечника — наиболее частая внепочечная аномалия; при этом разрыв дивертикула при аутосомно-доминантном поликистозе почек более вероятен, чем при дивертикулах, наблюдаемых в общей популяции.
Особенности фенотипических проявлений аутосомно-доминантного поликистоза почек (АДПКП):
• Возраст начала: от детства до зрелости
• Прогрессирующая почечная недостаточность
• Почечные и печеночные кисты
• Внутричерепные мешотчатые аневризмы
• Пролапс митрального клапана
• Дивертикулы толстого кишечника
Лечение аутосомно-доминантного поликистоза почек (АДПКП)
В основном аутосомно-доминантный поликистоз почек диагностируют по семейному анамнезу и УЗИ почек. Обнаружение почечных кист при УЗИ увеличивается с возрастом, так что 80-90% пациентов имеют обнаруживаемые кисты к 20 годам и почти 100% — к 30 годам жизни. Если это необходимо для пренатальной диагностики или идентификации родственника-донора почки, диагноз можно подтвердить анализом сцепления или прямым обнаружением мутации, или, в некоторых семьях, и тем, и другим.
Оказание помощи и лечение больных аутосомно-доминантным поликистозом почек нацелено на задержку развития почечной недостаточности и коррекцию симптомов. Гипертонию и инфекции мочевых путей следует энергично лечить, чтобы сохранить функцию почек. Боли, вызванные увеличением почек, уменьшаются при дренировании и склерозировании кист.
Риски наследования аутосомно-доминантного поликистоза почек
Приблизительно 90% пациентов имеют аутосомно-доминантный поликистоз почек в семейном анамнезе; только 10% аутосомно-доминантного поликистоза почек возникает вследствие новых мутаций в генах PKD1 или PKD2. Родители с аутосомно-доминантным поликистозом почек имеют 50% риск родить больного ребенка при каждой беременности. Если родители имели ребенка с внутриутробным началом болезни, риск родить другого сильно пораженного ребенка составляет приблизительно 25%. Тем не менее точно предсказать тяжесть проявлений болезни невозможно из-за варьирующей экспрессивности. Для семей, в которых известна мутация или возможен анализ сцепления, риск повторения может быть изменен при анализе ДНК плода.
Сибсы и родители пациентов с аутосомно-доминантным поликистозом почек также имеют повышенный риск болезни. Для обследования членов семьи рекомендуемый метод — ультрасонография почек.
Пример аутосомно-доминантного поликистоза почек (АДПКП). П.Д., 35-летний мужчина с пролапсом митрального клапана в анамнезе, поступает в местное отделение неотложной помощи с выраженными болями в боку и гематурией. Четыре месяца тому назад у него появились эпизодические боли в боку. УЗИ почек выявило нефролитиаз и множественные кисты почек, характерные для поликистоза почек. Данные его клинического осмотра в норме, за исключением систолического шума, соответствующего пролапсу митрального клапана, небольшой артериальной гипертензии и легкого повышения концентрации креатинина в сыворотке. Его отец и сестра умерли от прорыва внутричерепных аневризм, а сын умер в 1 год от поликистоза почек. После смерти сына врачи предлагали обследовать его и его жену на наличие поликистоза почек; однако родители решили не обследоваться из-за чувства вины и горя, вызванных смертью сына. Пациенту начато лечение камней в почках. Во время лечения нефролог сообщил больному, что у него аутосомно-доминантный поликистоз почек (АДПКП).
Поликистоз почек у детей
Поликистоз почек у детей – врожденная аномалия почек, характеризующаяся наличием в почечной ткани множественных мелкокистозных изменений, нарушающих функционирование органа. При манифестации поликистоза почек в детском возрасте отмечается стойкая высокая артериальная гипертензия, боли в пояснице, рецидивирующий пиелонефрит, почечная недостаточность. Диагноз поликистоза почек у детей подтверждается данными УЗИ почек, экскреторной урографии, ангиографии, сцинтиграфии и КТ почек. Лечение поликистоза почек у детей направлено на борьбу с ХПН; иногда проводится чрескожная пункция или лапароскопическое иссечение кист, трансплантация почки.
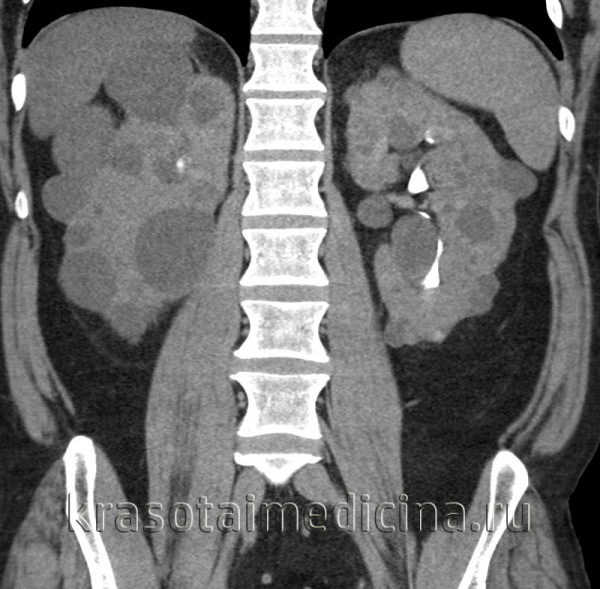
Поликистоз почек у детей (поликистозная болезнь, поликистозная дегенерация почек) – тяжелый структурный порок развития почек, при котором нормальная почечная ткань замещается множественными кистами различной величины. Поликистоз почек у детей встречается с частотой 1 случай на 250-1000 новорожденных, однако в связи с латентным течением в детском возрасте диагностируется редко. В зависимости от клинической формы, поликистоз почек может обнаружиться или проявиться в различные возрастные периоды: у новорожденных, детей раннего возраста, подростков или взрослых преимущественно в возрасте 30-40 лет. Кистозные изменения почек у детей нередко сочетаются с поликистозом печени, поджелудочной железы, селезенки, легких, поликистозными яичниками, мегауретером, добавочной почкой.
В связи с тем, что поликистоз почек у детей является наследственным заболеванием, его изучение является задачей урологии и генетики.
Классификация поликистоза почек
С учетом генетических аспектов выделят:
- поликистоз почек взрослых, тип I (мутация гена PKD1 в коротком плече 16-ой хромосомы)
- поликистоз почек взрослых, тип II (мутация гена РKD2 или РKD4 в 4-ой хромосоме)
- аутосомно-рецессивную поликистозную болезнь почек (мутация генов PKHD, ARPKD в 6-ой хромосоме). Данная форма поликистоза почек у детей сочетается с множественными пороками развития (расщелинами лица, врожденными пороками сердца и пр.).
- тяжелый инфантильный поликистоз почек с туберозным склерозом (мутация гена PKDTS в 16-ой хромосоме)
- врожденный микрокистоз почек финского типа (встречают у населения Финляндии и севера России)
- прочие разновидности поликистоза почек: поликистоз почек у детей в сочетании с катарактой и врожденной слепотой; поликистоз в сочетании микроцефалией, брахицефалией, гипертелоризмом и непропорционально короткими конечностями и др.
Поликистоз почек новорожденных наследуется по аутосомно-рецессивному типу. Поликистоз почек, наследуемый по аутосомно-доминантному типу, встречается у детей старшего возраста, подростков и взрослых.
В зависимости от степени кистозного поражения почек различают солитарные кисты, очаговую и тотальную форму поликистоза.
Причины поликистоза почек у детей
Формирование поликистоза почек у детей происходит уже в первые недели эмбриогенеза вследствие несрастания канальцев метанефроса и собирательных канальцев зачатка мочеточника. Согласно одной из гипотез, это может происходить вследствие иммунологической несовместимости метанефрогенной бластомы и мочеточникового ростка. Подтверждением этого предположения служит уменьшение в сыворотке пациентов с поликистозом концентрации С3-комплемента.
Кистозные полости, образующиеся из почечных канальцев, не соединенных с выводящей системой, могут быть гломерулярными, тубулярными и экскреторными. Кисты гломерулярного типа не связаны с канальцевой системой, поэтому не склонны к увеличению размеров. Данный тип характерен для поликистоза почек новорожденных и способствует раннему развитию хронической почечной недостаточности и смерти ребенка. Кисты тубулярного типа образуются из канальцев, а экскреторного типа - из собирательных трубочек. Увеличение кист происходит неравномерно, но постоянно из-за трудностей с опорожнением. Растущие кисты вызывают компрессию паренхимы и гибель значительной части нефронов.
Возникновение мутации генов, обусловливающих развитие поликистоза у детей, может быть вызвано воздействием химических и лекарственных веществ (консервантов продуктов, инсектицидов, препаратов лития, цитостатиков и пр.), вирусов (цитомегаловируса и т. д.) и другими неблагоприятными факторами.
Симптомы поликистоза почек у детей
Поликистоз почек новорожденных имеет злокачественное течение: нередко отмечается мертворождение, в остальных случаях летальный исход наступает первые месяцы или первый года жизни. Кистозное перерождение охватывает до 90% паренхимы почек. У новорожденных детей отмечается рвота, резкое увеличение почек и объемов живота; быстро нарастают признаки почечной недостаточности (увеличение в крови уровня мочевины и остаточного азота, олигурия и анурия), отеки, повышение АД. При этом гибель детей нередко наступает от сопутствующего респираторного дистресс-синдрома, обусловленного гипоплазией легких и пневмотораксом.
При поликистозе почек у детей старшего возраста большая часть почечных кист является открытыми; в почках сохраняется от 40 до 75% неповрежденной паренхимы. В связи с этим клиническая манифестация поликистоза почек у детей происходит позднее, во многом напоминая течение заболевания у взрослых. У детей обнаруживается увеличение размеров почек, гепатоспленомегалия, стойкая высокая артериальная гипертензия. Растяжение почечной ткани растущими кистами вызывает болевые ощущения в пояснице или в боку. Дети существенно отстают в росте, страдают анемией. Характерными осложнениями поликистоза почек у детей раннего и старшего возраста служат гематурия, мочекаменная болезнь, пиелонефриты, хроническая почечная недостаточность. Развитие печеночного фиброза приводит к портальной гипертензии, пищеводным и желудочно-кишечным кровотечениям. Гибель детей наступает от почечной или печеночной недостаточности через 2-15 лет от начала развития заболевания.
Диагностика поликистоза почек детей
Для постановки точного диагноза необходимо проведение полного клинико-лабораторного и инструментального обследования ребенка, анализ родословной. Дети с подозрением на поликистоз почек должны быть проконсультированы детским нефрологом (детским урологом) и генетиком.
Исследование общего анализа мочи при поликистозе почек у детей выявляет протеинурию, микро- или макрогематурию, лейкоцитурию (при пиелонефрите), гипо- или изостенурию. Для оценки функционального состояния почек проводится биохимическое исследование крови, исследование мочи по Зимницкому, проба Реберга.
Окончательное подтверждение и верификация диагноза осуществляется с помощью экскреторной урографии, почечной ангиографии, УЗИ, динамической сцинтиграфии, КТ почек и МР-урографии. Важнейшими признаками поликистоза почек у детей служат изменение размеров, контуров и расположения почек, деформация чашечек и лоханки, изменение сосудистой системы и др. При поликистозе почек детям может потребоваться УЗИ поджелудочной железы, печени, селезенки, яичников, поскольку нередко наблюдается кистозная трансформация этих органов.
Для выявления мутантного гена поликистоза почек у детей или родителей используются методы молекулярной гибридизации. На современном этапе поликистоз почек у ребенка может быть выявлен еще внутриутробно при скриннинговом акушерском УЗИ после 30-ой недели беременности.
Дифференциальную диагностику поликистоза почек у детей необходимо проводить с губчатой почкой, нефробластомой (опухолью Вильмса).
Лечение поликистоза почек детей
Терапия поликистоза почек у детей является пожизненной и направлена на предупреждение или борьбу с осложнениями, сохранение и улучшение функции почек. Дети с поликистозом почек нуждаются в повторных курсах медикаментозной терапии, постоянном соблюдении диетического и питьевого режима.
При сопутствующем воспалительном процессе в почках проводится курс лечения пиелонефрита антибиотиками и уросептиками. В случае присоединения артериальной гипертензии назначаются гипотензивные средства. При развитии ХПН на фоне поликистоза почек у детей проводится гемодиализ и ставится вопрос о трансплантации почки (иногда вместе с трансплантацией печени).
В случае быстрого увеличения кист в объеме может потребоваться хирургическое лечение: чрескожная пункционная аспирация кист под контролем УЗИ, лапароскопическое иссечение кист. При развитии портальной гипертензии может быть проведено портокавальное или спленоренальное шунтирование.
Прогноз при поликистозе почек детей
Прогноз при поликистозе почек у детей всегда серьезный; при этом, чем раньше заболевание начало прогрессировать, тем хуже. Младенческая форма поликистоза почек протекает крайне неблагоприятно и рано заканчивается гибелью детей. Практически у всех выживших детей к 20 годам формируется тяжелая почечная недостаточность. В этом случае единственным шансом на продление жизни больных является пересадка донорской почки.
Читайте также: