Генетика бета-талассемии. Наследование
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
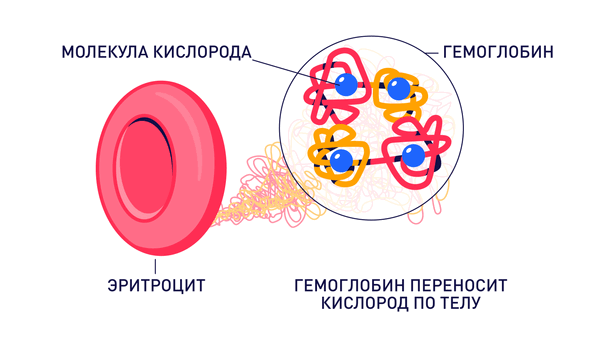
Талассемия – группа заболеваний наследственного характера, которые характеризуются нарушением синтеза определенных цепей гемоглобина. Сам гемоглобин состоит из двух частей: гем и глобин. Гем – это красящее вещество, небелковая часть состава, в которую входит железо. Глобин – это белковый компонент, который состоит из четырех белковых цепей: альфа-цепей и бета-цепей.
Внимание!
Здесь вы сможете выбрать врача, занимающегося лечением Талассемия Если вы не уверены в диагнозе, запишитесь на прием к терапевту или врачу общей практики для уточнения диагноза.
Статьи на тему Талассемия:
Симптомы
Причины
Диагностика
Лечение
Симптомы
При таком заболевании как талассемия, признаки могут быть различными. Наиболее часто проявляется талассемия у детей вместе со всеми симптомами. Характерны такие проявления:
- «башенная» (почти квадратная) форма черепа;
- сплющенная переносица;
- сужение щели глаз;
- увеличение верхней челюсти;
- увеличение селезенки и печени;
- бледность и пожелтение покровов кожи;
- язвы в зоне голеней;
- появление билирубиновых камней в путях желчи;
- высокая слабость и утомляемость;
- уменьшение сопротивляемости организма относительно стрессов;
- медленное физическое и половое развитие.
Формы
В зависимости от того, как поражаются цепи, выделяются такие формы заболевания:
- бета-талассемия (поражается бета-цепь);
- альфа-талассемия (поражены альфа-цепи):
- гетерозиготное носительство манифестного гена;
- гемоглобинопатия Н;
- альфа-талассемия гомозиготного типа;
В зависимости от формы различается:
- гомозиготная форма, которая считается результатом наследования гена от двух родителей;
- гетерозиготная форма, при которой ген передается посредством одного родителя.
- большая талассемия – крупная форма бета-талассемии, которая чревата такими осложнениями, как цирроз печени, трофические язвы, фиброз поджелудочной железы;
- промежуточная форма бета-талассемии. Протекает более доброкачественно. Признаки не так выражены, как в предыдущем варианте, и проявляются позже. Среди осложнений выделяется укрупнение селезенки и травмы костной системы;
- малая талассемия. Замечается только легкая анемия;
- минимальная бета-талассемия. Проходит без симптомов и обычно обнаруживается случайно.
В зависимости от степени заболевания выделяется:
- тяжелая форма (летальный исход еще в младенческом возрасте);
- хроническая форма средней тяжести (пациенты могут дожить до школьного возраста);
- хроническая форма легкой тяжести (больной доживает до взрослого возраста).
Причины
Причина заболевания – генная мутация того гена, который ответственен за синтез цепей глобина. Такой ген может быть унаследован от одного родителя или двух. Организм ребенка продуцирует цепи гемоглобина меньше или не продуцирует вовсе. Продукция других цепей, которые составляют глобин, при этом не заканчивается. Как результат, образуются белковые компоненты нестабильного характера, которые разрушают кровяные клетки. Потом проявляется анемия и во внутренних органах откладывается желез.
Диагностика
- Общий осмотр педиатром или гематологом на наличие характерных признаков: башенный череп, переносица седловой формы, узкие глаза, увеличенная печень и селезенка, бледность и пожелтение покровов кожи, язвы в области, отставание;
- также при такой болезни, как талассемия, анализ крови является необходимым методом диагностики: определяется снижение уровня гемоглобина, гипохромные эритроциты, показатель цвета, увеличение ретикулоцитов, повышение железа сыворотки;
- мазок крови: малый размер эритроцитов, изменение их формы;
- биохимия крови;
- электрофорез гемоглобина на ацетат-целлюлозной пленке;
- анализ биосинтеза цепей глобина;
- пункция костного мозга; костей;
- молекулярное исследование;
- исследования в лабораторных и клинических условиях;
- может понадобиться консультация гематолога и генетика.
Лечение
- В случае тяжелых форм показано переливание цельной крови или массы эритроцитов. Однако они дают только временный эффект и могут стать причиной осложнений;
- Сегодня самым эффективным считается переливание размороженных, отмытых либо фильтрованных эритроцитов, которые не так часто провоцируют побочные эффекты вместе с долгосрочным введением хелатов железа;
- Хелат железа нужно вводить длительно (на протяжении нескольких часов) под кожу. Потому созданы специальные препараты, которые можно присоединить к одежде. Больной, страдающий тяжелой формой заболевания, должен получать такой препарат на протяжении пяти дней в неделю всю свою жизнь. Чтобы предотвратить местное повреждение тканей, нужно иногда менять места инъекций. Если появляются гемолитические кризы, то необходимо введение небольших зон глюкокортикоидов;
- Если размеры селезенки увеличены, то ее удаляют. Такую операцию нельзя делать детям до 5 лет. Нормальный возраст – 8–10 лет. Позитивный эффект прослеживается на протяжении первого года после операции, а потом состояние может снова ухудшиться. Увеличивается риск инфекционных заболеваний;
- Сегодня оптимальным методом считается пересадка костного мозга. Это единственный радикальный метод лечения заболевания. Однако достаточно трудно найти донора;
- Больные должны соблюдать диету, которая вмещает большое количество танина: чай, какао, соя, орехи. Данные продукты помогают снизить всасывание железа.
Также есть необходимость в симптоматическом лечении:
- с целью улучшения печеночной функции назначаются гепопротекторы на протяжении не менее чем одного месяца;
- назначение витамина. С, который помогает выводить железо из организма.
Осложнения
Если форма заболевания тяжелая, то нередко появляются осложнения как результат отложения в тканях гемосидерина. Часто развивается цирроз печени и сахарный диабет, а также кардиосклероз, который может стать причиной смерти ребенка.
Талассемия: серьезные последствия небольшой мутации
Талассемия – тяжелое наследственное заболевание, возникающее из-за мутации генов гемоглобина. Этот белок заполняет эритроциты и связывает кислород, чтобы переносить его по крови. Когда белок перестает выполнять свою функцию из-за мутаций, организм подвержен серьезным нарушениям.
Содержание
Как проявляется талассемия?
- В первую очередь, для талассемии характерна тяжелая форма анемии – снижение количеств гемоглобина и эритроцитов и, следовательно, нарушение переноса кислорода и дыхания. Из-за анемии возникает слабость, головокружение, одышка.
- Кроме того, при талассемии организм нередко перегружен железом: ведь гемоглобина образуется меньше и атомы железа остаются в свободной форме в крови. Железо накапливается в органах и тканях и мешает им нормально функционировать. Прежде всего от этого страдают печень, сердце и эндокринные железы.
- Еще одно опасное последствие талассемии – увеличение размеров селезенки. Именно в этом органе происходит распад эритроцитов, который повышается при болезни. Чем больше селезенка, тем больше риск ее разрыва, который опасен для жизни пациентов.
- Также при талассемии часто деформируются кости. Это происходит потому, что из-за сниженного количества эритроцитов в костном мозге расширяются участки кроветворения. Клетки костного мозга даже могут выходить за пределы костей, образуя утолщения – псевдоопухоли. Это ведет не только к деформации скелета, но и к другим симптомам – ведь кости могут задевать нервы и сосуды.
![2021-09-22-Iron-Consumption.png]()
Как железо влияет на самочувствие и здоровьеМутации гемоглобина и тяжесть болезни
Гемоглобин состоит из двух типов цепей – альфа и бета. В соответствии с геном цепи, в котором произошла мутация, талассемии тоже делят на альфа- и бета-тип. При этом альфа-цепь гемоглобина у человека кодируют сразу две пары генов, а бета-цепь – всего одна. В зависимости от количества мутировавших аллелей, талассемии разделяют по тяжести.
![]()
Так, если перестает правильно работать лишь одна копия гена альфа-глобина, болезнь проходит почти бессимптомно. Нарушение работы двух копий ведет к легкой форме альфа-талассемии, трех – к клинически выраженным нарушениям (эта форма называется гемоглобинопатия Н). Если же неправильно работают все четыре копии гена альфа-глобина, чаще всего гибель происходит еще на этапе плода.
Похожим образом происходит и развитие бета-талассемии. Если одна из двух копий гена кодирует функциональный бета-глобин – талассемия проходит в легкой форме. При наличии в обеих копиях гена мутаций, влияющих на выработку бета-глобина, наблюдается тяжелая форма болезни – талассемия Кули.
А вот если нарушения происходят сразу и в альфа-, и в бета-цепи – образование гемоглобина компенсируется и болезнь проходит мягче. Дело в том, что негативный эффект мутаций в генах цепей гемоглобина состоит не только в снижении количества белка, переносящего кислород, но и в снижении равновесия между двумя типами цепей. Из-за этого поломка лишь в гене альфа-глобина может оказаться даже опаснее, чем сразу в двух типах генов. Однако, конечно, такие сочетания встречаются крайне редко.![2021-09-06-IDA.png]()
Железодефицитная анемия: кто в группе риска и как предотвратить болезнь?Наследование и эпидемиология
При этом в мире всего пять процентов населения – носители мутаций в генах цепей гемоглобина, а из них симптомам болезни подвержены чуть менее двух процентов людей. Стоит также учитывать, что жители разных регионов сталкиваются с талассемией с разной частотой. Прежде всего болезнь поражает жителей Средиземноморья, а также стран Центральной и Южной Азии. В России носителей мутаций, связанных с талассемией, сравнительно мало – около одного процента (для бета-типа).
Носители мутации могут даже не подозревать о своем статусе – часто повреждение лишь одной копии гена не ведёт к тяжелым симптомам. Однако носители мутации рискуют передать болезнь детям: если два носителя заведут ребенка, он может получить от родителей сразу обе копии мутировавшего гена с вероятностью около 25 процентов.
В предыдущем разделе мы уже обсуждали, что мутации в двух копиях генов приводит к более тяжелой форме талассемии. Но если посчитать вероятность такого события получится лишь около сотой процента.
Диагностика талассемии
Самый точный способ диагностировать талассемию любой тяжести – исследование структуры ДНК. Установление последовательности ДНК позволит обнаружить нарушенные варианты генов и не только вовремя принять меры в случае болезни, но и планировать беременность в случае бессимптомного носительства.
Талассемию также можно диагностировать при помощи анализов крови: общего, биохимического, а также определения в крови фракции гемоглобина. Эти исследования позволяют определить количество эритроцитов, их объем, а также концентрацию гемоглобина и уровни некоторых других веществ, чтобы по полученным результататам определить тип талассемии. При подозрении на талассемию также проводят УЗИ органов брюшной полости, чтобы оценить размеры селезенки и печени, которые страдают при болезни.
Генетический тест Атлас помогает определить патогенные варианты в гене, отвечающем за альфа-талассемию, обнаружить риски, связанные с проявлением симптомов и передачей заболевания детям.
Терапия и профилактика
Для лечения тяжелых форм талассемии используют переливание крови – для повышения количества эритроцитов и восстановления кислородного транспорта, а также хелатирование – для выведения свободной формы железа, которая не встроилась в «неправильный» гемоглобин. При тяжелой форме бета-талассемии переливание крови необходимо часто – каждые 3-4 недели. Однако сейчас такая терапия позволяет пациентам вести нормальный образ жизни и восстанавливает продолжительность жизни.
Для легких форм талассемии показания проще – врачи советуют соблюдать здоровый образ жизни и больше двигаться. Занятия спортом позволяют поддерживать минерализацию костей скелета и препятствуют их деформации.Генетика бета-талассемии. Наследование
Генетика бета-талассемии. Наследование
b-Талассемии имеют много общих характеристик с а-талассемией. Уменьшение синтеза b-глобина вызывает гипохромную микроцитарную анемию, а дисбаланс в синтезе глобинов ведет к осаждению избыточных а-цепей, вызывая патологию мембраны эритроцитов. В отличие от а-глобина, b-цепь важна только в послеродовом периоде. Следовательно, В-талассемии не проявляются несколько месяцев после рождения, когда b-глобин в норме заменяет у-глобин как основную не-a-цепь, и начинается синтез только основного гемоглобина взрослых, НbА.
Избыточные а-цепи накапливаются в эритроцитах и их предшественниках, приведя к их гибели и нарушению эритропоэза. Поскольку 6-ген остается неповрежденным, производство НbА2 продолжается, и, фактически, повышение уровня НbА2 уникально для гетерозиготных носителей b-талассемии. Уровень HbF также повышен, но не потому, что вновь активизировалась экспрессия гена у-глобина, отключенная при рождении, а из-за выборочного выживания или возможного повышения производства незначительной популяции эритроцитов взрослых, содержащих HbF.
В отличие от a-талассемий, b-талассемии обычно вызываются однонуклеотидными заменами, а не делециями. Во многих регионах мира, где часто встречается b-талассемия, имеется много разных мутаций гена b-талассемии, и наиболее вероятно, что лица, несущие два аллеля b-талассемии, окажутся генетическими компаундами, а не истинными гомозиготами по одному аллелю.
Большинство больных с двумя аллелями b-талассемии имеют «большую» талассемию, заболевание, проявляющееся выраженной анемией и пожизненной потребностью в медицинском наблюдении. Когда аллели b-талассемии допускают настолько малый синтез b-глобина, что гемоглобин полностью отсутствует, заболевание определяют как b0-талассемию. Если какое-то количество гемоглобина обнаруживается, говорят, что у пациента имеется b+-талассемия. Хотя тяжесть клинических проявлений болезни зависит от комбинированного эффекта двух имеющихся аллелей, до недавних пор доживание до взрослого возраста было редкостью.
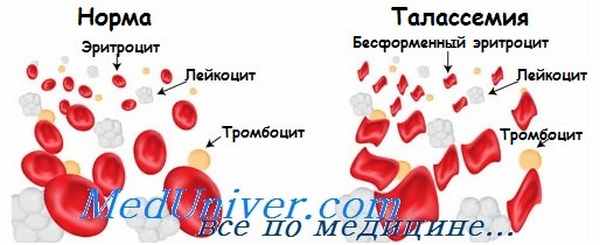
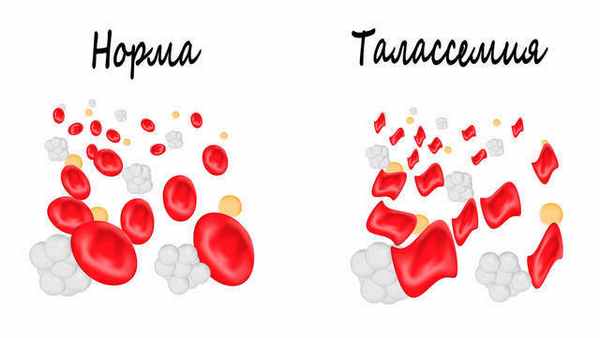
У детей, гомозиготных по b-талассемии, анемия развивается сразу же, как только уменьшается синтез HbF, обычно до 2 лет жизни. Эритроциты периферической крови заметно гипохромны и имеют различную величину и форму. В настоящее время лечение талассемий основано на коррекции анемии и стимуляции костного мозга переливаниями крови и стимуляции накопления железа назначением хе-латов. Пересадка костного мозга эффективна, но ее применяют только при наличии члена семьи с соответствующим HLA-генотипом.
Носители только одного аллеля b-талассемии клинически здоровы, такую форму заболевания называют «малой». Характерны гипохромные микроцитарные эритроциты, возможна легкая анемия, которая может быть диагностирована как железодефицитная. Диагноз малой талассемий подтверждается электрофорезом гемоглобинов, обычно показывающим увеличение уровня HbA2 (a2q2).
![бета-талассемия]()
b-Талассемия, сложные талассемий и наследственное персистирование фетального гемоглобина
Почти все известные типы мутаций, уменьшающих синтез мРНК или белка, описаны как причина b-талассемии. Таким образом, приведенный ниже обзор генетических дефектов, описывающих конкретные молекулярные основы одной из наиболее частых и тяжелых генетических болезней в мире, полезен для общего понимания мутационных механизмов. Мутации комплекса b-глобина подразделяют на две обширные группы с разными клиническими фенотипами. Одна группа дефектов, характерная для подавляющего большинства пациентов, нарушает синтез только В-глобина, вызывая простую b-талассемию.
Во второй группе мутаций большие делеции вызывают сложные талассемий, в которых утрачивается ген b-глобина, а также один или более других генов (или LCR) в группе b-глобина. Некоторые делеции в пределах группы b-глобина вызывают не талассемию, а довольно интересный фенотип, характеризующийся НПФГ (т.е. продолжение экспрессии гена у-глобина во взрослой жизни).
Молекулярная основа простой b-талассемии
Простая b-талассемия возникает вследствие множества различных типов молекулярных аномалий, преимущественно точковых мутаций, в гене b-глобина. Единственная частая делеция b-глобина среди всех расовых групп — 619-bр, частичная делеция 3'-конца гена у пациентов азиатского (индийского) происхождения.
Большинство мутаций, вызывающих простую b-талассемию, приводят к уменьшению количества синтезируемой мРНК b-глобина. Они включают мутации промотора, мутации сплайсинга РНК (наиболее частые), мутации, нарушающие фланкирование мРНК, и мутации сдвига рамки или нонсенс-мутации, вводящие кодон преждевременного завершения в пределах кодирующей области гена. Несколько структурных вариантов гемоглобина также нарушают процессинг мРНК b-глобина, например HbЕ, описанный ниже.
![бета-талассемия]()
Мутации сплайсинга РНК при бета-талассемии
Большинство пациентов с b-талассемиями, вызванными уменьшением синтеза мРНК b-глобина, имеют аномалии сплайсинга РНК. Описано более двух десятков дефектов этого типа, и, вместе взятые, они имеют большое клиническое значение. Эти мутации стали широко известны, поскольку их влияние на сплайсинг часто оказывалось неожиданно сложным, и анализ мутантных мРНК содействовал познанию критической последовательности нормального процессинга РНК. Дефекты сплайсинга разделяют на три группы, в зависимости от региона непроцессируемой РНК, в котором располагается мутация.
Группа 1, мутации сайта сплайсинга включает мутации в 5'-донорском или 3'-акцепторном участках интронов или в окружающих управляющих последовательностях. Критическая роль консервативных динуклеотидов GT в 5'-донорском участке и AG в З'-акцепторном участках интрона следует из полной утраты нормального сплайсинга, вызванной мутациями этих динуклеотидов. Инактивация нормального сайта акцептора приводит к использованию других последовательностей в исходной РНК, сходных с акцептором. Такие альтернативные сайты называют криптическими, поскольку в норме, при наличии правильного сайта, они не используются при сплайсинге. Криптические донорские или акцепторные сайты сплайсинга могут быть как в интронах, так и в экзонах, и могут использоваться в конкуренции с другими криптическими и нормальными сайтами сплайсинга.
Значение согласованных последовательностей, смежных с донорским или акцепторным динуклеотидами, также проявляется по эффектам мутаций. Например, замена пятого или шестого нуклеотида донорской последовательности интрона 1 уменьшает эффективность нормального сплайсинга, но, поскольку некоторый объем нормального сплайсинга все же происходит, формируется фенотип b+-талассемии.
Группа 2, мутации интрона — результат мутаций в пределах криптического сайта сплайсинга в интроне, повышающих использование этого сайта, что делает его более сходным или даже идентичным нормальному сайту сплайсинга. Затем пусковой криптический сайт с переменной эффективностью конкурирует с нормальным сайтом, уменьшая количество нормальной мРНК за счет нарушения сплайсинга в «правильном» сайте, остающемся вполне сохранным. Мутации криптических сайтов сплайсинга часто «неполные». Это означает, что происходит определенный объем сплайсинга с использованием нормального сайта, приводя к фенотипу b+-талассемии.
Группа 3, изменения кодирущей последовательности, влияющие на сплайсинг, вызываются мутациями сдвига рамки, которые активируют криптический сайт сплайсинга в экзоне. Например, одна из легких форм b+-талассемии вызвана мутацией в кодоне 24, активизирующей криптический сайт сплайсинга, но не изменяющей закодированную аминокислоту [как GGT, так и GGA кодируют глицин]; это пример синонимичной мутации, оказавшейся не безразличной по своему эффекту. Структурный вариант НbЕ демонстрирует, как нарушение сплайсинга и изменение в кодирующей последовательности могут вызываться единственной мутацией.
Нефункциональная мРНК при бета-талассемии
Некоторые синтезированные молекулы мРНК нефункциональны и не могут управлять синтезом полного полипептида, поскольку мутация генерирует преждевременные стоп-кодоны, раньше времени завершающие трансляцию белка. Две мутации около аминового конца поясняют этот эффект при b-талассемии. При одной (Gln39Stop) нарушение трансляции — следствие замены единственного нуклеотида, создающей нонсенс-мутацию. Другая мутация типа сдвига рамки происходит вследствие делеции одной пары оснований в начале открытой рамки считывания, с потерей первого нуклеотида 16 кодона, в норме кодирующего глицин; при произошедшем сдвиге рамки почти сразу образуется стоп-кодон, прерывающий дальнейшее считывание.
Поскольку синтез b-глобина отсутствует, оба типа нефункциональных мутаций мРНК вызывают b°-талассемию. В отличие от этого, мутации сдвига рамки, расположенные около карбоксильного конца белка, позволяют большой части мРНК транслироваться нормально или вызывают удлинение цепей глобина, чаще приводя к различным вариантам гемоглобина, а не к b°-талассемии.
Кроме снижения синтеза полипептида b-глобина, нонсенс-кодоны, включая два вышеописанных, часто приводят к уменьшению количества мутантной мРНК, которая на самом деле может не обнаруживаться. Механизмы, лежащие в основе этого феномена, названного нонсенс-опосредованным разрушением мРНК, не полностью понятны, но эффект ограничен нонсенс-кодонами, расположенными далее 50 пар оснований в 5'-конце последнего соединения между экзонами.
Дефекты фланкирующих последовательностей мРНК бета-глобина при бета-талассемии
Две мутации при b+-талассемии указывают на критическую роль посттранскрипционных модификаций любой мРНК, кэпирования РНК, ограничивающее 5'-конец, и полиаденилирования 3-конца мРНК. Обнаружен один пациент с заменой аденина на цитозин в первом нуклеотиде мРНК (это место — сайт кэпирования — в 90% эукариотической мРНК — пурин).
Мутация приводит к нарушению формирования кэпа, вызывая распад РНК. Полиаденилирование мРНК происходит после ферментативного расщепления, а сигнал точки расщепления, AAUAAA, находится около 3'-конца в большинстве эукариотической мРНК. Пациент с мутацией, изменяющей сигнальную последовательность на ААСААА, синтезировал только незначительное количество мРНК b-глобина, полиаденилированной в нормальном положении.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Талассемии
(средиземноморская анемия; большая и малая талассемия)
, MD, PhD, Johns Hopkins University School of Medicine
- Патофизиология
- Клинические проявления
- Диагностика
- Прогноз
- Лечение
- Основные положения
- Дополнительная информация
Талассемии – это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского, средиземноморского, или южноазиатского происхождения. Бета-талассемия более распространена у лиц средиземноморского, ближневосточного, южноазиатского и индийского происхождения. Симптомы и признаки обусловлены анемией, гемолизом, спленомегалией, гиперплазией костного мозга, при многократных гемотрансфузиях может наблюдаться перегрузка железом. Диагностика основана на генетическом исследовании и количественном анализе структуры гемоглобина. Лечение тяжелых форм может включать в себя гемотрансфузии, спленэктомию, терапии хелаторами и трансплантацию стволовых клеток.
Патофизиология талассемий
Талассемия – гемоглобинопатия Обзор гемоглобинопатий Гемоглобинопатии – это генетические нарушения, влияющие на структуру или синтез молекулы гемоглобина. Молекулы гемоглобина состоят из полипептидных цепей, химическая структура которых контролируется. Прочитайте дополнительные сведения , которая является одним из наиболее распространенных наследственных заболеваний, связанных с синтезом гемоглобина. Нормальная зрелая молекула гемоглобина (гемоглобин А) состоит из 2 пар цепей, называемых альфа и бета. Нормальная кровь взрослого человека также содержит ≤ 2,5% Hb A2 (состоит из альфа- и дельта-цепей) и 2% гемоглобина F (фетального гемоглобина), который имеет гамма-цепи вместо бета-цепей. Талассемия является результатом несбалансированного синтеза гемоглобина, вызванного снижением синтеза по крайней мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта).
Альфа-талассемия
Альфа-талассемия является результатом снижения синтеза альфа-полипептидных цепей вследствие делеции одного или нескольких генов альфа-цепей. Люди, как правило, имеют четыре гена альфа-цепей (по два на каждой паре хромосом), потому что ген альфа-цепей дублируется. Классификация болезни основана на количестве и местоположении делеций:
Альфа + талассемия: Потеря одного гена на одной хромосоме (альфа/--)
Альфа 0 талассемии: Потеря обоих генов на одной и той же хромосоме (--/--)
Бета-талассемия
Бета-талассемия вызвана снижением синтеза бета-полипептидных цепей в результате либо мутации, либо делеции в гене бета-глобина, что приводит к нарушению синтеза гемоглобина А. Мутации или делеции могут привести к частичной потере (бета + аллель) или полной потере (бета 0 аллель) функции бета-глобина. Существуют два гена бета-глобина, и у пациентов могут быть гетерозиготные, гомозиготные или сложные гетерозиготные мутации. Кроме того, пациенты могут быть гетерозиготными или гомозиготными по аномалиям в 2-х различных генах глобина (например, бета и дельта).
Бета-дельта-талассемия является менее распространенной формой бета-талассемии, при которой нарушается синтез как дельта-цепи, так и бета-цепи. Эти мутации могут быть гетерозиготными или гомозиготными.
Симптомы и признаки талассемий
Клинические особенности талассемий сходны, но различаются по степени тяжести, в зависимости от количества нормального гемоглобина.
Альфа-талассемия
Пациенты с одной альфа + аллелью (альфа/альфа; альфа/--) являются клинически нормальными и называются бессимптомными носителями.
У гетерозигот с дефектами в 2 из 4 генов, таких как две альфа + аллели (альфа/--; альфа/--) или 1 альфа 0 аллель (альфа/альфа; --/--) наблюдается тенденция к развитию микроцитарной анемии легкой или умеренной степени тяжести, но с субклиническим течением. Данные пациенты имеют малую альфа-талассемию.
Дефекты в 3 из 4 генов, вызванные совместным наследованием как альфа +, так и альфа 0 (альфа/-; -/-), существенно нарушают синтез альфа-цепи. Синтез поврежденных альфа-цепей в результате приводит к образованию тетрамеров избыточных бета-цепей, называемых Hb H, а у младенцев - к формированию гамма-цепей, называемых гемоглобином Барта. У пациентов с болезнью гемоглобина Н часто наблюдаются гемолитическая анемия и спленомегалия.
Дефект всех 4 генов через две альфа 0 аллели (--/--;--/--) является летальным состоянием, которое вызывает внутриутробную гибель плода (водянку плода), поскольку гемоглобин, не содержащий альфа-цепей, не переносит кислород.
Бета-талассемия
При бета-талассемии, клинические фенотипы подразделяются на 3 группы в зависимости от степени нарушения синтеза бета-глобина:
Малая бета-талассемия (характерно) возникает у обычно бессимптомных гетерозигот (бета/бета + бета/бета 0) с клинической картиной микроцитарной анемии от легкой до умеренной степени. Этот фенотип может также возникнуть в легких случаях бета +/бета +.
Промежуточная бета-талассемия проявляется вариабельной клинической картиной, которая является промежуточной между большой и малой талассемией, обусловленная наследованием 2 аллелей бета-талассемии (бета +/бета 0 или, в тяжелых случаях, бета +/бета +).
Большая бета-талассемия (или анемия Кули) возникает у гомозиготных пациентов (бета 0/бета 0) или сложных гетерозигот (бета 0/бета +) в результате тяжелого дефекта бета-глобина. У этих пациентов развивается тяжелая анемия и гиперактивность костного мозга. Большая бета-талассемия проявляется в возрасте от 1 до 2 лет с симптомами тяжелой анемии и трансфузионной и абсорбционной перегрузки железа. У пациентов наблюдаются желтуха, язвы нижних конечностей и холелитиаз (как при серповидноклеточной анемии Серповидно-клеточная анемия Серповидноклеточная анемия ( гемоглобинопатия) является причиной хронической гемолитической анемии, которая наблюдается практически исключительно у представителей негроидной расы. Она вызвана. Прочитайте дополнительные сведенияДиагностика талассемий
При наличии подозрения диагностика данного типа гемолитической анемии включает в себя:
Мазок периферической крови
Исследование структуры ДНК (пренатальная диагностика)
Малую талассемии обычно выявляют при проведении рутинного мазка периферической крови и развернутого анализа крови, когда обнаруживают микроцитарную анемию и повышенное количество эритроцитов. При желании, диагноз малой бета-талассемии может быть подтвержден с помощью количественных исследований структуры гемоглобина. Никакого вмешательства не требуется; у женщин анемия может усугубляться беременностью.
Более тяжелую талассемию следует подозревать у пациентов с отягощенным наследственным анамнезом, при наличии характерных клинических симптомов или микроцитарной гемолитической анемии. При подозрении на талассемию выполняются обычные лабораторные тесты для выявления микроцитарных гемолитических анемий и количественный анализ структуры гемоглобина. Характерно повышение уровня билирубина, железа и ферритина в сыворотке крови.
При альфа-талассемиях процентное содержание Hb F и Hb A2 обычно в пределах нормы, выявление одного или двух дефектных генов, характерных для талассемии, производится с помощью современных генетических тестов. Диагноз часто ставится путём исключения других причин микроцитарной анемии.
При большой бета-талассемии наблюдается тяжелая анемия, часто со снижением уровня гемоглобина ≤ 6 г/дл (≤ 60 гр/л). Количество эритроцитов повышено по отношению к уровню гемоглобина, поскольку наблюдается выраженный микроцитоз. Диагностика основана на исследовании мазка периферической крови, в котором обнаруживается множество ядросодержащих эритробластов, мишеневидных клеток, небольших бледноокрашенных эритроцитов; характерна точечная или диффузная базофилия.
При количественном анализе структуры гемоглобина диагностическим критерием малой бета-талассемии является повышенный уровень Hb A2. При большой бета-талассемии обычно повышено содержание Hb F, иногда до 90%, а содержание Hb A2 обычно > 3%.
Болезнь гемоглобина Н диагностируется по выявлению Hb H или фракций Барта при электрофорезе гемоглобина. Наличие специфического молекулярного дефекта не меняет клинического подхода.
Стандартом пренатальной диагностики и генетического консультирования является картирование генов рекомбинантной ДНК (в частности метод полимеразной цепной реакции [ПЦР]).
Если при анемии выполняется исследование костного мозга (к примеру, для исключения других причин), выявляется выраженная эритроидная гиперплазия.
При визуальном исследовании, выполненном по другим причинам, у пациентов с большой бета-талассемией можно выявить изменения, обусловленные хронической гиперактивностью костного мозга. Наблюдаются истончение кортикального слоя костей черепа, расширение диплоических пространств, лучистая трабекулярная структура, появление гранул или феномен «матового стекла». В трубчатых костях могут наблюдаться очаги остеопороза, расширение костномозгового пространства и истончение кортикального слоя. Тела позвонков могут иметь зернистый внешний вид, или вид матового стекла. Фаланги могут быть прямоугольными или двояковыпуклыми. Визуализация грудной клетки может выявить признаки паравертебрального экстрамедуллярного гемопоэза.
Прогноз при талассемии
Продолжительность жизни у пациентов с малой бета-талассемией или малой альфа-талассемией является нормальной. Прогноз при заболевании Hb Н и промежуточной бета-талассемии вариабельный.
Продолжительность жизни снижена у пациентов с большой бета-талассемией, главным образом в связи с осложнениями в результате хронических трансфузий.
Лечение талассемий
Часто – переливание эритроцитарной массы с/без железохелатирующей терапии
Спленэктомия при наличии спленомегалии
Если возможно, выполняют трансплантацию аллогенных стволовых клеток
Луспатерцепт для лечения трансфузионно-зависимой бета-талассемии
Пациентам с малой альфа-талассемией или малой бета-талассемией лечение не требуется.
При гемоглобинозе Н: спленэктомия может помочь при тяжелой анемии или наличии спленомегалии.
Пациенты с промежуточной бета-талассемией должны получать как можно меньше гемотрансфузий, чтобы избежать перегрузки железом. Тем не менее супрессия патологического гемопоэза с помощью периодических трансфузий эритроцитов Эритроциты (ККТ) Цельная кровь способствует улучшению кислородной емкости крови, расширению объема и замене факторов свертывания, а раньше ее назначали при сильной кровопотере. Однако, поскольку компонентная. Прочитайте дополнительные сведения может быть ценной у пациентов с тяжелым течением заболевания. При большой бета-талассемии следует проводить трансфузии по необходимости для поддержания уровня гемоглобина примерно 9 - 10 г/дл (от 90 до 100 г/л) и избегать развития тяжелых клинических проявлений.
Чтобы предотвратить или отсрочить развитие осложнений от перегрузки железом, должен быть удален избыток (трансфузионного) железа (например, путем назначения длительной железохелаторной терапии Лечение Вторичная перегрузка железом появляется в результате избыточной абсорбции железа, повторяющихся переливаниях крови или избытке перорального приема, как правило, у пациентов с нарушениями эритропоэза. Прочитайте дополнительные сведения ). Хелаторную терапию, как правило, начинают, когда уровни сывороточного ферритина превышают 1000 нг/мл (> 1000 мкг/л) или после приблизительно от 1 до 2 лет проведения плановых трансфузий. Спленэктомия может снизить потребность в гемотрансфузиях у пациентов с выраженной спленомегалией.
Люспатерцепт представляет собой инъекционный рекомбинантный белок слияния, который ингибирует метаболический путь передачи сигналов от трансформирующего фактора роста бета. В рандомизированном плацебо-контролируемом исследовании у пациентов с бета-талассемией он снизил потребность в трансфузии на 33% у 21% пациентов (по сравнению с 4,5% в контрольной группе). Луспатерцепт является вариантом лечения для пациентов, которым необходимо переливание крови ( 1 Справочные материалы по лечению Талассемии – это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского. Прочитайте дополнительные сведения ).
Справочные материалы по лечению
1. Cappellini MD, Viprakasat V, Taher A, et al: A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med 382(13):1219–1231, 2020. doi: 10.1056/NEJMoa1910182
Основные положения
Талассемия является результатом снижения синтеза по меньшей мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта); в результате аномальные эритроциты являются микроцитарными, часто неправильной формы и склонны к гемолизу (вызывающему анемию).
Часто проявляется спленомегалия, часто массивная, которая может привести к селезеночной секвестрации, ускоряющей разрушение эритроцитов (в том числе введенных при переливании).
Часто наблюдается перегрузка железом из-за увеличенного поглощения (в связи с нарушенным эритропоэзом), а также из-за частых трансфузий.
Диагностика с использованием электрофореза гемоглобина.
Следует проводить трансфузии по необходимости, проводя мониторинг перегрузки железом и используя хелаторную терапию.
Спленэктомия может снизить потребность в гемотрансфузиях у пациентов со спленомегалией.
Трансплантация аллогенных стволовых клеток имеет излечивающий эффект.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Cooley's Anemia Foundation: provides comprehensive patient education and support and advocacy to patients with thalassemia
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Талассемия
![Талассемия]()
Талассемия, в буквальном переводе "морская анемия", возникает она при мутации генов, отвечающих за синтез пептидов, из которых состоит гемоглобин, и тяжесть заболевания зависит от числа изменённых генов. Особенность этой анемии в том, что пациенту приходится бороться с избыточным потреблением железа.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” - это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Коды МКБ-10 (Международная классификация болезней 10 пересмотра):
- альфа-талассемия - D56.0
- бета-талассемия - D56.1
- дельта-бета-талассемия - D56.2
- носительство признака талассемии - D56.3
- наследственное персистирование фетального гемоглобина (НПФГ) - D56.4
- другие талассемии - D56.8
- талассемия неуточненная - D56.9
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников - бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия - увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Правильный образ жизни при заболевании
Существуют простые советы, которые помогают людям с талассемией лучше переносить симптомы заболевания.
- не употреблять никакие витаминно-минеральные комплексы или пищевые добавки, содержащие железо;
- сбалансированное разнообразное питание;
- профилактика инфекционных заболеваний, в первую очередь вакцинация.
Прогноз при талассемии
При легкой форме талассемии прогноз очень хороший. Такие пациенты не требуют постоянного лечения, и осложнения крайне редки. При средней и тяжелой форме прогноз также хороший, но необходимы регулярные переливания крови и прием препаратов, связывающих железо. Такие лекарства очень важны, поскольку наибольшее количество смертей больных талассемией связано с накоплением этого элемента в организме. Пациентам с костными осложнениями может понадобиться операция.
Профилактика талассемии
Талассемия – заболевание наследственное. Поэтому, если один или оба родителя больны, необходимо проконсультироваться с врачом-генетиком на этапе планирования беременности.
Читайте также: