Генетика несовершенного остеогенеза. Наследование
Добавил пользователь Morpheus Обновлено: 27.01.2026
Несовершенный остеогенез (НО) – это группа генетически обусловленных заболеваний, в основе которых лежит нарушение формирования костной ткани. В свою очередь, избыток тиреоидных гормонов при болезни Грейвса (БГ) оказывает негативное влияние на костную ткань, тем самым усугубляя течение НО, что требует от эндокринолога максимально внимательного ведения пациентов с сочетанием данных патологий. В настоящей статье представлен уникальный клинический случай сочетания БГ и НО.
Ключевые слова
Для цитирования:
For citation:
Введение
Несовершенный остеогенез (НО, лат. – osteogenesis imperfecta) – гетерогенная группа наследственных заболеваний соединительной ткани и скелета, характеризующаяся повышенной частотой переломов костей и другими полиорганными осложнениями, ассоциированная с нарушением синтеза коллагена I типа.
Распространенность НО в популяции составляет 1 на 10 000–20 000 новорожденных.
Патогенез НО в большинстве случаев обусловлен нарушением синтеза коллагена I типа, который является основным компонентом органического матрикса костной ткани. НО может быть обусловлен либо недостаточным синтезом коллагена I типа, либо образованием дефектного коллагена в результате нарушений этапов формирования третичной структуры белка. Также при НО изменяется баланс между резорбцией и формированием кости в сторону преобладания резорбции. В результате этого возникают деформации костей и увеличивается риск переломов [1].
Основными клиническими характеристиками пациентов с НО являются переломы костей от воздействия таких некритичных по травматичности факторов, как изменение положения тела, бег, ходьба, кашель/чихание и пр. Слабость костной ткани при НО варьирует по тяжести: от низкой костной массы до прогрессирующих деформаций скелета с высокой частотой переломов. У пациентов с НО могут иметь место задержка физического развития, сколиоз, прогрессирующие деформации длинных костей, тугоухость, патология прорезывания зубов (dentinogenesis imperfecta), голубые склеры, черепно-лицевые аномалии, гипермобильность суставов и дыхательная недостаточность [2, 3]. Необычный окрас склер объясняется тем, что сквозь особенно прозрачную, истонченную склеру просвечивает пигмент сосудистой оболочки. Также существует мнение, что голубой цвет склеры является не следствием ее истончения, а повышения прозрачности, что объясняется изменениями в коллоидно-химических качествах ткани. На этом основании предлагается более верный для обозначения подобного патологического состояния термин, который звучит как «прозрачная склера».
Большинство случаев (до 90%) НО имеют аутосомно-доминантный тип наследования с семейным мозаицизмом из-за мутаций в генах COL1A1 или COL1A2, кодирующих коллаген I типа [3]. Примерно в 10% случаев НО может проявляться при рецессивном аллеле мутантного гена [2].
Впервые мутация в гене COL1A2 у пациента с летальной формой НО была выявлена в 1983 г. [4]. В настоящее время описано более 1500 мутаций, детерминирующих НО [5].
В настоящее время нет единой классификации НО, а различные способы ранжирования на типы и подтипы основаны либо исключительно на клинических проявлениях, либо на клинико-рентгенологических проявлениях и варианте генетического дефекта. Наиболее часто используется классификация по Sillence, дополненная M. Ramachandran, которая включает в себя данные клинического и рентгенологического обследования пациента и позволяет выделить четыре клинико-генетических типа заболевания (табл. 1). В классификации M. Ramachandran принимается во внимание состояние дентиногенеза (табл. 2). По этому критерию принципиально выделяют тип В, который сопровождается нарушениями дентиногенеза, и подтип А, который таковыми не проявляется.
Таблица 1. Классификация несовершенного остеогенеза по Sillence D.O. (1979; расширение в 1984 г.)
I – АД тип с голубыми склерами
В различной степени ломкость костей, голубые склеры, ранняя потеря слуха, незначительное отставание линейного роста
I-A: нормальные зубы
I-B и I-C: несовершенный дентиногенез
II – перинатально летальная форма рентгенологически характеризуется тяжелыми саблевидно-варусными деформациями бедренных костей и переломами ребер
Крайне высокая ломкость костей, перинатальная смерть
II-A: короткие и широкие длинные трубчатые кости с переломами, широкие ребра с переломами.
II-B: короткие и широкие длинные трубчатые кости с переломами, широкие ребра с единичными переломами
II-C: тонкие длинные трубчатые кости с переломами, тонкие ребра
III – прогрессивно-деформирующий тип, с нормальными склерами
От средней до тяжелой степени ломкость костей, голубые склеры в младенчестве
Раннее начало кифосколиоза
Несовершенный дентиногенез может быть
IV- АД с нормальным цветом склер
Ломкость костей, от средней до тяжелой степени деформации длинных трубчатых костей и позвоночника, нормальный цвет склер, от незначительного до тяжелой степени отставание линейного роста
IV-A: нормальные зубы
IV-B: несовершенный дентиногенез
Таблица 2. Классификация несовершенного остеогенеза по Ramachandran M. (2008)
у 20% кифоз или кифосколиоз
наличие вормиевых (вставочных) костей
АД, спонтанные мутации, семейный мозаицизм
наличие вормиевых костей с отсутствием оссификации
АД, редко АР, семейный мозаицизм
прогрессирующие деформации длинных костей, позвоночника
голубые при рождении и белые у взрослых
гипопластические вормиевы кости
инвалидность, чаще на инвалидной коляске
гипопластические вормиевы кости
АД, семейный мозаицизм
Поражение костной системы при НО является патогномоничным признаком, но отнюдь не единственным, поскольку коллаген содержится практически во всех органах и тканях. Далеко не все из них проявляются клинически: чаще всего описаны респираторные и сердечно-сосудистые нарушения. Мы представляем уникальный клинический случай сочетания НО и болезни Грейвса (БГ).
Клинический случай
В отдел радионуклидной диагностики и терапии ФГБУ ЭНЦ поступила молодая девушка 19 лет с диагнозом диффузного токсического зоба (БГ) на лечение радиоактивным йодом.
- 04.2013 – манифестация тиреотоксикоза, по поводу которого назначен постоянный прием (без осложнений) тиреостатиков (тирозол) по схеме «блокируй» в максимальной дозе 40 мг с последующим снижением до минимальной поддерживающей дозы 7,5 мг;
- 09.2016 направлена на радиойодтерапию (РЙТ) в связи с неэффективностью длительной (38 мес) терапии тиреостатиками, рецидивами тиреотоксикоза на фоне снижения/отмены препарата.
Результаты анализа крови при обследовании в отделении: ТТГ – 0,001 мМЕ/л (0,2–4,0), св.Т4 – 26,9 пмоль/л (9–20), св.Т3 – 7,8 пмоль/л (2,5–5,5), уровень антител к рецептору ТТГ – 7,2 МЕ/л.
Сцинтиграфия щитовидной железы с Тс-99м-пертехнетатом – общий захвата технеция – 13,9% (0,3–1,7%), признаки гиперфункции щитовидной железы (рис. 1). По УЗИ объем щитовидной железы 46,3 мл (норма для женщин: до 18 мл), эхографические признаки аутоиммунного тиреоидита, васкуляризация при цветном дуплексном картировании (ЦДК) диффузно усилена, узловых образований нет.
Рис. 1. Сцинтиграфия щитовидной железы с Tc99.
При клиническом осмотре обращено внимание на голубой оттенок склер глаз (рис. 2) и гипермобильность суставов. Из анамнеза установлено, что пациентка с 3-летнего возраста до настоящего времени перенесла многократные переломы костей скелета, которые происходили при незначительных травмах: левая большеберцовая кость, правая большеберцовая кость, левое колено, левая лучевая кость, правое колено, кости черепа. Последний перелом произошел в январе 2015 г. В связи с диагностированным остеопорозом на основании низкой минеральной плотности костной ткани (МПКТ) и низкотравматических переломов получала терапию антирезорбтивными препаратами: ибандронат 150 мг внутрь в течение 2 мес, с 2014 г. получила 4 инъекции деносумаба (Пролиа) 60 мг п/к каждые 6 мес (последняя инъекция в августе 2016 г.). На фоне лечения пациентка отмечала уменьшение частоты переломов. Денситометрия позвоночника от июня 2016 г. (рис. 3): по Z-критерию в позвоночнике -3,4SD, в бедре -2,8SD, шейке бедра – 1,9 SD. В динамике на фоне лечения деносумабом и препаратами кальция в декабре 2016 г. отмечена значимая положительная динамика с повышением МПКТ по Z-критерию в позвоночнике до -1,3 SD, в бедре - до -1,5 SD, в шейке бедра – до -0,3 SD. При оценке трабекулярного костного индекса TBS (рис. 4) в области позвоночника данных за значимое нарушение архитектоники трабекул в области скелета не получено.
Рис. 2. Синий оттенок склер пациентки.
Рис. 3. Денситометрия позвоночника от пациентки.
Рис. 4. Оценка трабекулярного костного индекса пациентки.
Лабораторная оценка кальциевого гомеостаза при поступлении: Сa общий – 2,36 ммоль /л (2,15–2,55), Са ионизированный – 1,32 ммоль/л (1,03–1,32), паратгормон – 38 пг/мл (15–65), щелочная фосфатаза – 58 ЕД/л (0–270), остеокальцин – 17,53 нг/мл (11–35), витамин D (общий 25(ОН)D) – 26,9 нг/мл (30–100), бета-кросслапс – 0,061 нг/мл (<0,580).
Таким образом, результаты исследований свидетельствуют о компенсации кальций-фосфорного обмена при небольшой недостаточности витамина D. Снижение бета-кросслапс в нижний диапазон нормы у молодой девушки является следствием подавления резорбции костной ткани под действием деносумаба.
Проведено медико-генетическое обследование.
Ребенок от близкородственного брака: биологические родители – двоюродные сибсы.
На этапе подготовки к РЙТ произведено дозиметрическое планирование путем изучения индивидуальных параметров радиобиокинетики после перорального назначения трейсерной активности 131 (2 МБк): эффективное время полувыведения 131 из организма составило 139 ч, интегральный захват радиофармпрепарата (РФП) – 49%, время максимального накопления – 9 ч, рекомендуемая лечебная активность 131 – 1100 МБк.
11.11.2016 выполнена РЙТ 131I пероральной активностью 1100 МБк (29,7 мКи): лечение прошло без осложнений, выписана с «закрытого» режима на 4-е сутки (остаточный фон на расстоянии 1 м. от тела: 15,9 мкЗв/ч) с персональными рекомендациями по радиационной безопасности.
Рис. 5. Область шеи до проведения радиойодтерапии.
Рис. 6. Область шеи через 3 мес после радиойодтерапии.
Больной был назначен план динамического наблюдения, результаты которого (в настоящее время прошло 4 мес после РЙТ) представлены в таблице 3.
Таблица 3. Результаты исследований после проведения радиойодтерапии
Описанный клинический случай сочетания НО и БГ является уникальным. В медицинской литературе нам удалось обнаружить только одно клиническое наблюдение сочетания НО с клиническими проявлениями тиреотоксикоза (снижение массы тела, повышение уровня метаболизма, повышение температуры тела, тахикардия, тахипноэ) и повышением уровня тироксина в крови, опубликованное в 1972 г. Cropp G.J. и соавт. [7]. Имевшиеся у пациента метаболические изменения исчезли в начале пубертата. В то время НО как патологический процесс был недостаточно изучен. Авторы предположили, что изменения костной ткани были преходящими и индуцированы собственно БГ [3].
Дефект коллагена сам по себе не объясняет повышения тиреоидных гормонов, но, по предположению Diegidio P. и соавт. [8], дефектная соединительная ткань может приводить к снижению эффективности естественного барьера между тканью щитовидной железы и кровотоком, «оголяя» таким образом антигены клеток щитовидной железы для иммунной системы, которая в нормальных условиях о них не знает и не обладает к ним толерантностью, что и запускает аутоиммунную реакцию [3].
Избыток тиреоидных гормонов негативно влияет на костную ткань. Это обусловлено тем, что Т3 влияет как на остеобласты, так и на остеокласты. Тиреотоксикоз характеризуется активацией костного обмена (резорбции и костеобразования), что вследствие более длительного времени процессов костеобразования приводит суммарно к супрафизиологическому повышению потери костной ткани. Логично предположить, что сочетание НО и тиреотоксикоза БГ у нашей пациентки усугубляло течение первичной остеопатии НО.
Тиреотоксикоз является общепризнанной причиной вторичного остеопороза, при котором активация резорбции кости может приводить даже к тяжелой гиперкальциемии [9]. Высокие уровни свободных тиреоидных гормонов напрямую активируют формирование остеокластов, что приводит к повышению резорбции костной ткани. Частота гиперкальциемии у пациентов с тиреотоксикозом до 60 лет – 2,3% и пациентов старше 60 лет – 18,8% [10].
Эффективным лечением по профилактике переломов при НО и тиреотоксикозе являются антирезорбтивные препараты: бисфосфонаты и деносумаб. Подавление резорбции при НО увеличивает костную массу, улучшает размеры и форму позвонков у пациентов, получающих лечение, в сравнении с пациентами без лечения [11, 12].
В настоящее время все больше обсуждается превосходство деносумаба как в лечении остеопороза, так и других метаболических болезней скелета. Это обусловлено тем, что бисфосфонаты, однажды примененные для лечения, будут связаны с костями в течение многих лет, что ставит большие вопросы о безопасности их использования у детей, подростков и молодых лиц репродуктивного возраста. Деносумаб (Пролиа), полностью человеческое антитело IgG2, которое связывается с RANK-лигандом, был одобрен в 2010 г. для лечения остеопороза у женщин в постменопаузе. Препятствуя взаимодействию RANK-лиганда с его RANK-рецептором, деносумаб является мощным антирезорбтивным агентом, снижающим дифференцировку и выживаемость остеокластов и, следовательно, подавляющим резорбцию кости [12].
При использовании деносумаба в лечении детей с НО значимо увеличивается МПКТ и рост пациентов [13]. При этом рентгенографически в сравнении с бисфосфонатами отмечается формирование более плотной кости, с отсутствием «линий зебры» (чередование полос неравномерно плотной, после применения бисфосфонатов, и разреженной кости), что отражает более равномерное формирование костной ткани [14]. Эти данные позволяют сделать заключение об имеющемся возможном преимуществе деносумаба над бисфосфонатами у пациентов с НО.
Лечение БГ у нашей пациентки проводилось по стандартному подходу. Вначале лечение тиреостатиками, которое было эффективно, однако за его время не произошло регресса аутоиммунной агрессии против щитовидной железы, что, с учетом личного предпочтения пациентки, послужило основанием предложить ей дальнейшее лечение РЙТ. Нами проводилось стандартное дозиметрическое планирование. Примечательна высокая эффективность проведенной РЙТ в виде быстрого и выраженного регресса объема щитовидной железы (в 5 раз), а также сравнительно быстрого купирования тиреотоксикоза с переходом в недостаточность гормонов щитовидной железы. Можно предположить, что такая эффективность связана также с нарушенной структурой коллагена и таким образом большей подверженностью тиреоцитов к действию РФП. Но при этом повышения частоты побочных реакций не наблюдалось.
Заключение
Представленный клинический случай обосновывает оценку тиреоидной функции у пациентов с НО с целью исключения тиреотоксикоза как фактора, усугубляющего данный вид остеопатии и ее возможных осложнений. Демонстрирует высокую эффективность РЙТ в лечении БГ данной пациентки. Суждение о влиянии НО на эффективность РЙТ и планирование дозы РФП требует дальнейшего изучения.
Дополнительная информация
Источник финансирования
Обследование пациента проведено за счет средств ФГБУ «Эндокринологический научный центр» Минздрава России.
Согласие пациента
Пациентка добровольно подписала информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Ожирение и метаболизм».
Конфликт интересов
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Современная классификация и молекулярно-генетические аспекты незавершенного остеогенеза
Незавершенный остеогенез (несовершенный остеогенез в русскоязычной литературе) – наиболее распространенная наследственная форма ломкости костей, генетически и клинически гетерогенное заболевание с широким спектром клинической тяжести, основное клиническое проявление которого – множественные переломы начиная с натального периода жизни, зачастую приводящие к инвалидизации с детского возраста. К основным клиническим признакам незавершенного остеогенеза относятся голубые склеры, потеря слуха, аномалия дентина, повышенная ломкость костей, нарушение роста и осанки с развитием характерных инвалидизирующих деформаций костей и сопутствующих проблем, включающих дыхательные, неврологические, сердечные, почечные нарушения. Незавершенный остеогенез встречается и у мужчин, и у женщин, заболевание наследуется как по аутосомно-доминантному, так и аутосомно-рецессивному типам, существуют спорадические случаи заболевания, обусловленные мутациями de novo, а также обнаружены Х-сцепленные формы. Термин «незавершенный остеогенез» был введен W. Vrolick в 1840-х гг. Первая классификация заболевания сделана в 1979 г. и неоднократно пересматривалась из-за идентификации молекулярной причины заболевания и открытия новых механизмов развития незавершенного остеогенеза. В начале 1980-х гг. мутации в двух генах коллагена типа I (COL1A1и COL1A2) впервые были ассоциированы с аутосомно-доминантным типом наследования незавершенного остеогенеза. С тех пор идентифицированы еще 18 генов, продукты которых участвуют в процессах формирования и минерализации костной ткани. До сих пор не определена степень генетической гетерогенности заболевания, исследователи продолжают идентифицировать новые гены, вовлеченные в его патогенез, число которых достигло 20. В последнее десятилетие стало известно, что аутосомно-рецессивные, аутосомно-доминантные и Х-связанные мутации в широком спектре генов, кодирующих белки, участвующие в синтезе коллагена типа I, его процессинге, секреции и посттрансляционной модификации, а также в белках, регулирующих дифференцировку и активность костеобразующих клеток, вызывают несовершенный остеогенез. Большое количество причинных генов усложнило классическую классификацию заболевания, и в связи с новыми достижениями в области молекулярных основ незавершенного остеогенеза постоянно совершенствуется и классификация. В этом обзоре мы систематизировали и обобщили информацию о результатах исследований в области изучения клинико-генетических аспектов незавершенного остеогенеза и отразили современное состояние классификационных критериев диагностики заболевания.
Ключевые слова
Об авторах
Институт биохимии и генетики – обособленное структурное подразделение Уфимского федерального исследовательского центра Российской академии наук
Россия
Уфа
Институт биохимии и генетики – обособленное структурное подразделение Уфимского федерального исследовательского центра Российской академии наук; Республиканский медико-генетический центр
Россия
Уфа
Список литературы
1. Asharani P.V., Keupp K., Semler O., Wang W., Li Y., Thiele H., Yigit G., Pohl E., Becker J., Frommolt P., Sonntag C., Altmüller J., Zimmermann K., Greenspan D.S., Akarsu N.A., Netzer C., Schönau E., Wirth R., Hammerschmidt M., Nürnberg P., Wollnik B., Carney T.J. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am. J. Hum. Genet. 2012;90(4):661-674. DOI 10.1016/j.ajhg.2012.02.026.
2. Barnes A.M., Cabral W.A., Weis M., Makareeva E., Merta E.L., Leikin S., Eyre D., Trujillo C., Marini J.C. Absence of FKBP10in recessive type XI OI leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 2012;33(11):1589-1598. DOI 10.1002/humu.22139.
4. Cabral W.A., Chang W., Barnes A.M., Weis M.A., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J., Bulas D.I., Kozma C., Smith P.A., Eyre D.R., Marini J.C. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39(3):359-365. DOI 10.1038/ng1968.
5. Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet.2010;86(3):389-398. DOI 10.1016/j.ajhg.2010.01.034.
8. Dwan K., Phillipi C.A., Steiner R.D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst. Rev. 2014;7:Cd005088.
9. Fratzl-ZelmanN., Barnes A.M., Weis M., Carter E., Hefferan T.E., Perino G., Chang W., Smith P.A., Roschger P., Klaushofer K., Glorieux F.H., Eyre D.R., Raggio C., Rauch F., Marini J.C. Non-lethal type VIII osteogenesis imperfecta has elevated bone matrix mineralization. J. Clin. Endocrinol. Metab. 2016;101(9):3516-3525. DOI 10.1210/jc.2016-1334.
10. Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., Lalic L., Glorieux D.F., Fassier F., Bishop N.J. Type V osteogenesis imperfecta: a new form of brittle bone disease. J. Bone Miner. Res. 2000;15(9):1650-1658. DOI 10.1359/jbmr.2000.15.9.1650.
11. Glorieux F.H., Ward L.M., Rauch F., Lalic L., Roughley P.J., Travers R. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002;17(1):30-38. DOI 10.1359/jbmr.2002.17.1.30.
12. Grafe I., Alexander S., Yang T., Lietman C., Homan E.P., Munivez E., Chen Y., Jiang M.M., Bertin T., Dawson B., Asuncion F., Ke H.Z., Ominsky M.S., Lee B. Sclerostin antibody treatment improves the bone phenotype of Crtap (–/–) mice, a model of recessive Osteogenesis Imperfecta. J. Bone Miner. Res. 2016;31(5):1030-1040.
13. Hoyer-Kuhn H., Franklin J., Allo G., Kron M., Netzer C., Eysel P., Hero B., Schoenau E., Semler O. Safety and efficacy of denosumab in children with osteogenesis imperfect – a first prospective trial. J. Musculoskelet Neuronal. Interact.2016;16(1):24-32.
14. Ignatovich O.N., Namazova-Baranova L.S., Margieva T.V., Yakhyaeva G.T., Zhurkova N.V., Savostyanov K.V., Pushkov A.A., Krotov I.A. Osteogenesis imperfecta: diagnostic feature. Pediatricheskaya Pharmacologiya = Pediatric Pharmacology. 2018;15(3): 224-232. DOI 10.15690/pf.v15i3.1902. (in Russian)
16. Laine C.M., Joeng K.S., Campeau P.M., Kiviranta R., Tarkkonen K., Grover M., Lu J.T., Pekkinen M., Wessman M., Heino T.J., Nieminen-Pihala V., Aronen M., Laine T., Kröger H., Cole W.G., Lehesjoki A.E., Nevarez L., Krakow D., Curry C.J., Cohn D.H., Gibbs R.A., Lee B.H., Mäkitie O. WNT1mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013; 368:1809-1816. DOI 10.1056/NEJMoa1215458.
17. Lapunzina P., Aglan M., Temtamy S., Caparrós-Martin J.A., Valencia M., Letón R., Martinez-Glez V., Elhossini R., Arm K., Vilaboa N., Ruiz-Perez V.L. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet.2010;87(1):110-114. DOI 10.1016/j.ajhg.2010.05.016.
18. Li L., Zhao D., Zheng W., Wang O., Jiang Y., Xia W., Xing X., Li M. A novel missense mutation in P4HB causes mild osteogenesis imperfecta. Biosci. Rep. 2019;39(4). DOI 10.1042/BSR20182118.
19. Lindahl K., Langdahl B., Ljunggren O., Kindmark A. Treatment of osteogenesis imperfecta in adults. Eur. J. Endocrinol.2014;171(2): R79-R90. DOI 10.1530/EJE-14-0017.
20. Lindert U., Cabral W.A., Ausavarat S., Tongkobpetch S. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X linked osteogenesis imperfecta. Nat. Commun. 2016;7:11920. DOI 10.1038/ncomms11920.
21. Lowenstein E.J. Osteogenesis imperfecta in a 3,000-year-old mummy. Childs Nerv. Syst.2009;25(5):515-516. DOI 10.1007/s00381-009-0817-7.
23. Marini J.C., Forlino A., Bächinger H.P., Bishop N.J., Byers P.H., De Paepe A., Fassier F., Fratzl-Zelman N., Kozloff K.M., Krakow D., Montpetit K., Semler O. Osteogenesis imperfecta. Nat. Rev. Dis. Primers. 2017;3:1-19. DOI 10.1038/nrdp.2017.52.
24. Mendoza-Londono R., Fahiminiya S., Majewski J. Care4Rare Canada Consortium; Tétreault M., Nadaf J., Kannu P., Sochett E., Howard A., Stimec J., Dupuis L., Roschger P., Klaushofer K., Palomo T., Ouellet J., Al-Jallad H., Mort J.S., Moffatt P., Boudko S., Bächinger H.P., Rauch F. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet.2015;96(6): 979-985. DOI 10.1016/j.ajhg.2015.04.021.
25. Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J., Bachinger H.P., Pace J.M., Schwarze U., Byers P.H., Weis M.A., Fernandes R.J., Eyre D.R., Yao Z., Boyce B.F., Lee B. CRTAPis required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127(2):291-304. DOI 10.1016/j.cell.2006.08.039.
27. Pigarova E.A., Sheremeta M.S., Kulikova K.S., Belovalova I.M., Tulpakov A.N., Rumiantsev P.O. Osteogenesis imperfecta in combination with Graves disease. Ozhirenie i Metabolism = Obesity and Metabolism. 2017;14(4):77-82. DOI 10.14341/OMET2017477-82. (in Russian)
28. Puig-Hervás M.T., Temtamy S., Aglan M., Valencia M., MartínezGlez V., Ballesta-Martínez M.J., López-González V., Ashour A.M., Amr K., Pulido V., Guillén-Navarro E., Lapunzina P., CaparrósMartín J.A., Ruiz-Perez V.L. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome-osteogenesis imperfecta phenotypic spectrum. Hum. Mutat. 2012;33(10):1444-1449. DOI 10.1002/humu.22133.
29. Pyott S.M., Tran T.T., Leistritz D.F., Pepin M.G., Mendelsohn N.J., Temme R.T., Fernandez B.A., Elsayed S.M., Elsobky E., Verma I., Nair S., Turner E.H., Smith J.D., Jarvik G.P., Byers P.H. WNT1mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet.2013;92(4): 590-597. DOI 10.1016/j.ajhg.2013.02.009.
31. Rubinato E., Morgan A., D’Eustacchio A., Pecile V., Gortani G., Gasparini P. A novel deletion mutation involving TMEM38Bin a patient with autosomal recessive osteogenesis imperfecta. Gene.2014; 545(2):290-292. DOI 10.1016/j.gene.2014.05.028.
32. Sinder B.P., Salemi J.D., Ominsky M.S., Caird M.S., Marini J.C., Kozloff K.M. Rapidly growing Brtl/+ mouse model of osteogenesis imperfecta improves bone mass and strength with sclerostin antibody treatment. Bone. 2015;71:115-123.
34. Tournis S., Dede A.D. Osteogenesis imperfecta – a clinical update. Metabolism. 2018;80:27-37. DOI 10.1016/j.metabol.2017.06.001.
35. VanDijk F.S., Nesbitt I.M., Zwikstra E.H., Nikkels P.G.J., Piersma S.R., Fratantoni S.A., Jimenez C.R., Huizer M., Morsman A.C., Cobben J.M., van Roij M.H.H., Elting M.W., Verbeke M.I.J.L., Wijnaendts L.C.D., Shaw N.J., Högler W., McKeown C., Sistermans E.A., Dalton A., Meijers-Jeijboer H., Pals G. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet.2009; 85(4):521-527. DOI 10.1016/j.ajhg.2009.09.001.
36. Ward L.M., Rauch F., Travers R., Chabot G., Azouz E.M., Lalic L., Roughley P.J., Glorieux F.H. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone.2002;31(1): 12-18.
37. Weaver C.M., Alexander D.D., Boushey C.J., Dawson-Hughes B., Lappe J.M., LeBoff M.S., Liu S., Looker A.C., Wallace T.C., Wang D.D. Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the National Osteoporosis Foundation. Osteoporos. Int. 2016;27:367-376.
38. Yakhyayeva G.T., Margieva T.V., Namazova-Baranova L.S., Savostyanov K.V., Pushkov A.А., Zhurkova N.V., Zherdev K.V., Vashakmadze N.D., Gevorkyan A.K. Clinical case of rare type V osteogenesis imperfecta. Pediatricheskaya Pharmakologiya = Pediatric Pharmacology. 2015a;12(1):79-84. (in Russian)
39. Yakhyayeva G.T., Namazova-Baranova L.S., Margieva T.V. New aspects of the genetic basis, classification and treatment of osteoge nesis imperfecta: a literature review. Pediatricheskaya Pharmakolo giya = Pediatric Pharmacology. 2015b;12(5):579-588. DOI 10.15690/pf.v12i5.1461. (in Russian)
Генетика несовершенного остеогенеза. Наследование
Генетика несовершенного остеогенеза. Наследование
Несовершенный остеогенез — группа наследственных заболеваний, предрасполагающих к переломам костей, даже вследствие небольшой травмы, и к скелетным нарушениям. Обнаружен значительный размах степени клинических проявлений, от летальной перинатальной формы до легкого увеличения частоты переломов.
Около 90% больных имеют мутации в двух генах, COL1A1 и COL1A2, кодирующих цепи коллагена I типа, основного белка костей. Клиническая гетерогенность, по крайней мере частично, может объясняться локусной и аллельной гетерогенностью; фенотипы изменяются в зависимости от того, какая цепь проколлагена I типа затронута, а также от типа и положения мутации в локусе.
Кроме того, при некоторых формах имеется другая область локусов первичных мутаций. Общая встречаемость всех форм болезни почти 1 на 15 000.
Нормальная структура коллагена в связи с несовершенным остеогенезом
Чтобы понимать патогенез несовершенного остеогенеза, нужно знать основные характеристики коллагена I типа в норме. Коллаген I типа — основной структурный белок костных и других волокнистых тканей. Молекула проколлагена I типа формируется из двух proаl(I) цепей (кодируемых в хромосоме 17 геном COL1A1) и одной аналогичной, но отличающейся цепи proа2(1) (кодируемой в хромосоме 7 геном COL1A2).
Белки, формирующиеся из субъединиц, подобные коллагену, часто подвергаются мутациям, которые повреждают связывание субъединиц, изменяя их точки взаимодействия. Тройная спираль коллагена формируется 338 последовательно расположенными повторами Gly-X-Y; в позиции X часто оказывается пролин, а в позиции Y — гидроксипро-лин или гидроксилизин.
Глицин, самая маленькая из аминокислот, — единственный достаточно компактный остаток, способный занять осевое положение внутри спирали, поэтому мутации, заменяющие какие-либо остатки на глицин, разрушительно действуют на третичную структуру белка.
Несколько особенностей созревания проколлагена играют особую роль в патофизиологии несовершенного остеогенеза. Во-первых, сборка отдельных proa цепей в тример начинается с карбоксильного конца, и образование тройной спирали продолжается по направлению к аминовому концу.
Следовательно, мутации, изменяющие остатки в карбоксильном конце тримера, более разрушительные, поскольку они создают помехи на более ранних этапах формирования тройной спирали. Во-вторых, посттрансляционная модификация (например, гидроксилирование пролина или лизина; гликозилирование) проколлагена продолжается в частях цепи, не собравшихся в тройную спираль.
Таким образом, если сборка спирали снижается вследствие мутаций, несобранные части дефектных цепей избыточно модифицируются, что замедляет их переход во внеклеточное пространство.
Избыточная модификация также может создавать помехи образованию коллагеновых фибрилл. В результате всех этих аномалий число молекул коллагена уменьшается, при этом многие из них аномальны. В костях аномальные цепи и их уменьшенное количество приводят к нарушению минерализации фибрилл коллагена.
Молекулярные аномалии коллагена при несовершенном остеогенезе
У больных с несовершенным остеогенезом обнаружено более 800 различных мутаций, влияющих на синтез или структуру коллагена I типа. Клиническая гетерогенность болезни отражает даже большую гетерогенность на молекулярном уровне. Мутации входят в два общих класса, уменьшающих синтез проколлагена I типа и изменяющих структуру собранных молекул. Теперь стало возможным в известной мере предсказывать фенотип, вызываемый специфическим типом молекулярного дефекта.
Тип I несовершенного остеогенеза: снижение синтеза коллагена I типа. Большинство индивидуумов с несовершенным остеогенезом I типа имеют мутации, приводящие к синтезу клетками примерно половины нормального количества проколлагена I типа. Наиболее часто это результат мутаций, образующих преждевременный стоп-кодон в одном аллеле COL1A1, которые делают мРНК этого аллеля очень неустойчивой.
Поскольку молекулы проколлагена I типа для формирования должны иметь две pro-альфа-l(I) цепи, недостаток половины количества мРНК приводит к синтезу половины нормального количества молекул проколлагена I типа, хотя эти молекулы нормальны. Миссенс-мутации вызывают более легкую форму несовершенного остеогенеза, если заменяемая аминокислота располагается в аминовом конце молекулы, поскольку замены в этой позиции менее разрушительны для сборки цепи коллагена.
Тип II, III и IV несовершенного остеогенеза: структурно дефектные коллагены. Фенотипы несовершенного остеогенеза II, III и IV типов вызваны мутациями, приводящими к структурно аномальной pro-альфа-l цепи; замены в pro-альфа-2 цепи оказывают сравнимый эффект. Чаще всего эти пациенты имеют в тройной спирали замены глицина более крупным остатком.
Специфика повреждения коллагена, позиция замены, природа заменяющего остатка — все это крайне важные фенотипические детерминанты, тем не менее возможно сделать некоторые обобщения о влиянии на фенотип специфических аминокислотных замен. Например, замены в pro-альфа-l (I) цепи более распространены у пациентов с несовершенным остеогенезом III и IV типа и чаще летальны. В обеих цепях замена глицина (нейтральный остаток) на аспартат (кислый остаток) обычно очень разрушительна и гораздо чаще связана с тяжелым фенотипом (типа II).
Иногда специфическая замена оказывается связанной с более чем одним фенотипом, что, вероятно, отражает влияние мощных генов-модификаторов данного моногенного заболевания.
Новые формы несовершенного остеогенеза, не вызванные мутациями коллагена. В течение последних нескольких лет выделены три дополнительных формы несовершенного остеогенеза (тип V, VI и VII), не связанных с мутациями в гене коллагена I типа. Мутантные гены не найдены, хотя локус несовершенного остеогенеза VII типа картирован на коротком плече хромосомы 3 и наследуется как рецессивный признак. Другие формы наследуются доминантно и имеют отличающиеся клинические характеристики или патологию костей, но все они похожи на несовершенный остеогенез IV типа.
Генетика несовершенного остеогенеза
Большинство мутаций в гене коллагена I типа, вызывающих несовершенный остеогенез, действуют доминантно, но есть и несколько рецессивных. По крайней мере некоторые механизмы возникновения различных типов наследования при разных мутациях в одной молекуле обнаружены при изучении биохимических дефектов. В целом эта болезнь иллюстрирует генетические сложности, возникающие, когда мутации изменяют структурные белки, особенно сформированные из многочисленных различных субъединиц.
Сравнительно мягкий фенотип и доминантное наследование несовершенного остеогенеза I типа объясняется тем, что хотя синтезируется только половина необходимого количества молекул, они нормального строения. Более серьезные последствия синтеза структурно дефектных pro-альфа-l (I) цепей (по сравнению с отсутствием синтеза любых цепей) частично отражают стехиометрию коллагена I типа, содержащего две proal цепи и одну pro-альфа-2 цепь.
Соответственно если половина pro-альфа-1 (I) цепей аномальна, три из четырех молекул содержат по крайней мере одну аномальную цепь; в отличие от этого, если дефектна половина pro-альфа-2(I) цепей, будет затронута только одна из двух молекул. Таким образом, мутации типа миссенс-аллеля proal (I) (pro-альфа-lM) — доминантные отрицательные аллели, поскольку они нарушают как pro-альфа-l, так и pro-альфа-2 цепи.
Другими словами, эффект мутантного аллеля увеличивается из-за полимерной природы молекулы коллагена. Следовательно, при доминантно наследуемых болезнях типа несовершенного остеогенеза лучше иметь мутацию, приводящую к полному отсутствию продукта гена, чем к синтезу аномального продукта.
Хотя мутации, приводящие к структурным аномалиям pro-альфа-2 цепей, лишь вдвое уменьшают число нормальных молекул коллагена I типа (против трех четвертей при структурно аномальных цепях pro-альфа-l), это уменьшение, тем не менее, достаточно, в случае некоторых мутаций, чтобы вызвать тяжелый перинатальный летальный фенотип.
Большинство младенцев со II типом несовершенного остеогенеза, перинатальной летальной формой, имеют новую доминантную мутацию, и, следовательно, вероятность повторения в семье очень низкая. В отдельных случаях, тем не менее, несовершенным остеогенезом II поражается более одного сибса. Такие повторения обычно вызваны родительским мозаицизмом. Достоверно документированных случаев аутосомно-рецессивных форм несовершенного остеогенеза II типа не представлено, но описано несколько примеров рецессивного наследования несовершенного остеогенеза III типа.
Лечение и пренатальная диагностика несовершенного остеогенеза
Если молекулярный дефект у пациента может быть определен, возрастающие знания соответствия между генотипом и фенотипом несовершенного остеогенеза дают возможность в известной мере предсказывать течение болезни.
Кроме того, установление факта, что дефект наследуется от больного родителя (аутосомно-доминантного), здорового родителя (с половым мозаицизмом), двух здоровых гетерозиготных родителей (аутосомно-рецессивных) или как новая мутация, позволяет рассчитать точный риск повторения.
Пренатальная диагностика при несовершенном остеогенезе II типа (перинатальная летальная форма) может выполняться обследованием черепа и длины конечностей при УЗИ во II триместре беременности. При беременности с высоким риском для пренатальной диагностики необходим анализ коллагена, который синтезируют клетки, культивированные из биоптатов ворсин хориона, или прямой анализ мутации, заранее идентифицированной в семье.
Хотя лечение несовершенного остеогенеза ограничено общими медицинскими и хирургическими мерами, эта ситуация изменяется из-за открытия, что лекарства класса бифосфонатов, уменьшая резорбцию костной ткани, могут увеличивать костную плотность у некоторых больных. Более существенный вопрос, уменьшают ли бифосфонаты частоту и тяжесть переломов при несовершенном остеогенезе, исследуется, но такая перспектива выглядит многообещающей.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Несовершенный остеогенез
Несовершенный остеогенез является наследственным нарушением коллагена, вызывающим диффузную ненормальную хрупкость костей и иногда сопровождающимся нейросенсорной тугоухостью, голубыми склерами, несовершенным дентиногенезом и гипермобильностью суставов. Диагноз обычно ставится на основе клинических данных. Лечения включает применение гормона роста для некоторых типов и бисфосфонатов.
Выделяют 4 основных типа несовершенного остеогенеза:
У 90% людей, которые имеют один из этих основных типов, обнаруживают мутации в генах, кодирующих про-альфа-цепи проколлагена 1-го типа (структурный компонент костей, связок и сухожилий), COL1A1 или COL1A2. Другие типы встречаются редко и вызываются мутациями иных генов.
Симптомы и признаки несовершенного остеогенеза
Потеря слуха присутствует у 50–65% пациентов с незавершенным остеогенезом и может возникать при любом из 4 типов.
Тип I самый легкий. Симптомы и признаки у некоторых пациентов ограничены синими склерами (из-за дефицита в соединительной ткани коллагена, позволяющего нижележащим сосудам просвечивать через нее) и костно-мышечной болью из-за гипермобильности суставов. В детстве возможны повторяющиеся переломы.
Тип II (неонатальный летальный тип или врожденный несовершенный остеогенез) является наиболее тяжелой, летальной формой. Множественные врожденные переломы приводят к формированию укороченных конечностей. Склеры синие. Череп мягкий и при пальпации ощущается как мешок с костями. Поскольку череп мягкий, травмы во время родов могут привести к внутричерепным кровоизлияниям и мертворождению или новорожденный может внезапно умереть в течение первых нескольких дней или недель жизни.
Тип III является наиболее тяжелой формой несмертельного несовершенного остеогенеза. Пациенты с типом III имеют низкий рост, искривление позвоночника, а также множественные рецидивирующие переломы. Макроцефалия с треугольным лицом и деформации грудной клетки являются общими симптомами. Оттенок склер варьируется.
Тип IV – промежуточная степень тяжести. Выживаемость высока. Кости легко ломаются в детстве до подросткового возраста. Склеры, как правило, нормального цвета. Рост умеренно низкий. Точный диагноз очень важен, поскольку этим пациентам лечение помогает.
Диагностика несовершенного остеогенеза
В некоторых случаях, анализ проколлагена I типа или генетическое исследование
Диагноз незавершенного остеогенеза обычно основывается на клинических данных, но стандартные критерии отсутствуют.
При неопределенном клиническом диагнозе можно провести анализ проколлагена 1-го типа из культуры фибробластов (из биоптатов кожи) или анализ последовательности для генов COL1A1 и COL1A2.
Тяжелые формы несовершенного остеогенеза могут быть обнаружены внутриутробно при помощи УЗИ 2-го уровня.
Лечение несовершенного остеогенеза
Гормон роста помогает детям с проблемным ростом (типы I и IV).
Лечение бисфосфонатами направлено на повышение плотности костной ткани, уменьшение боли в костях и риска переломов ( 1 Справочные материалы по лечению Несовершенный остеогенез является наследственным нарушением коллагена, вызывающим диффузную ненормальную хрупкость костей и иногда сопровождающимся нейросенсорной тугоухостью, голубыми склерами. Прочитайте дополнительные сведенияОртопедическая хирургия, физиотерапия и трудотерапия помогают предотвратить переломы и улучшить функцию.
Кохлеарная имплантация показана в отдельных случаях при потере слуха.
Справочные материалы по лечению
1. Dwan K, Phillipi CA, Steiner RD, Basel D: Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev CD005088, 2016. 10.1002/14651858.CD005088.pub4
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Osteogenesis Imperfecta (OI) Foundation: Организация, предоставляющая поддержку, образование и научную информацию о НО
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Памятка о несовершенном остеогенезе

Если в семье рождается «хрупкий» ребёнок, родителям приходится непросто, и сразу возникает огромное количество вопросов. Что делать? Как помочь малышу? Как сделать так, чтобы он рос как можно более самостоятельным и подвижным? Ответы на эти вопросы дали мамы наших подопечных Ольга Коршунова и Светлана Акбарова, которые собрали полезную информацию и написали памятку для тех, кто впервые столкнулся с врождённой хрупкостью костей.
Инструкция для тех, кто впервые столкнулся с несовершенным остеогенезом (НО)
Самое главное:
Разделы памятки
I. Общая информация
Несовершенный остеогенез (НО) — наследственная дисплазия соединительной ткани, для которой характерны хрупкость костей и деформации конечностей. В 85% случаев мутации возникают в генах COL1A1 и COL1A2, что приводит к количественным и качественным изменениям синтеза коллагена первого типа. Несовершенный остеогенез нельзя вылечить полностью. Лечение может быть только симптоматическим, и это очень непростая задача, требующая комплексного мультидисциплинарного подхода. Основное направление лекарственной терапии — применение бисфосфонатов, которые повышают минеральную плотность костей. Хирургическое лечение переломов и деформаций конечностей улучшает качество жизни, хотя и сопровождается частыми осложнениями. Реабилитационная терапия играет огромную роль в восстановлении двигательной активности «хрупких» людей после переломов и операций.
Типы несовершенного остеогенеза
Течение болезни может сильно различаться. Она может протекать легко, а может привести к летальному исходу ещё до рождения ребёнка или сразу после него. Типы заболевания в зависимости от тяжести его течения распределены так: I < IV < III < II, и всё-таки существуют данные о том, что симптомы и тяжесть НО могут сильно различаться даже в пределах одного типа и одной семьи. На данный момент в клинической практике чаще всего используется классификация заболевания по Sillence:
I тип — самая лёгкая форма, для неё характерны частые переломы, голубые склеры и нарушение слуха. Переломы проявляются в раннем возрасте, когда ребёнок начинает ходить, их частота снижается после завершения роста. Деформации конечностей возникают редко, часто у пациентов нормальный рост. Несовершенный дентиногенез (так называемые опаловые зубы) встречается редко.
II тип — перинатально летальная форма с наиболее тяжёлыми проявлениями, если ребенку удается выжить в родах. Множественные переломы выявляют уже на внутриутробном этапе. Конечности ребёнка обычно короткие и имеют дугообразные деформации. Цвет склер (белков глаз) голубой или сероватый.
III тип — характерны прогрессирующие деформации конечностей. Форма лица часто треугольная, с выступающими лобными буграми, склеры голубого или сероватого цвета. Иногда встречаются осложнения, требующие консультации и лечения у нейрохирургов.
IV тип — средней степени тяжести. Для типа характерны несовершенный дентиногенез (так называемые опаловые зубы), нарушение слуха, вариабельность роста. Иногда встречаются осложнения, требующие консультации и лечения у нейрохирургов.
V тип — особенность заключается в формировании гипертрофической костной мозоли и оссификации (окостенении) межкостной мембраны на предплечье.
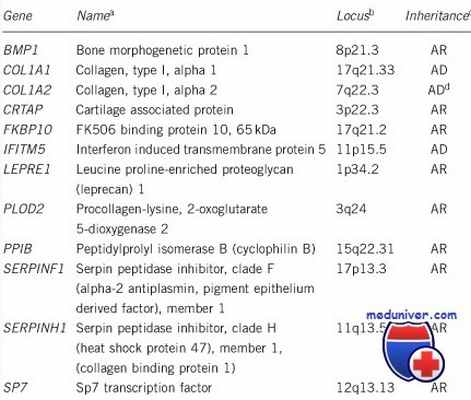
По мере развития генетики были обнаружены другие гены, отвечающие за развитие НО. Каждому новому гену был присвоен новый тип (см. Таблицы 1–3 ).
Генетическая классификация несовершенного остеогенеза (1)
| Мутировавший | Белок | Тип | Путь наследования | Клинические особенности |
| Нарушения синтеза и структуры коллагена | ||||
| COLIA1 COLIA2 | a1 (COLIA1) и a2 (COLIA2) коллаген | I, II, III, IV | АД | Классические фенотипы, описанные D. Sillence |
| Нарушение минерализации кости | ||||
| IFITM5 | (BRIL) Bone-restricted interferon induced transmembrane protein (IFM5) | V | АД | Тяжесть скелетных деформаций варьируется от отсутствия до тяжёлых форм, цвет склер — от нормального до синего, возможны оссификация межкостных мембран, вывих головки лучевой кости, потеря слуха |
| SERPINF1 | (PEDF) Pigment epithelium-derived factor | VI | АР | Тяжесть скелетных деформаций варьируется от средней до тяжёлой: наличие остеоида, чешуеподобное строение кости |
| Нарушения посттрансляционной модификации коллагена | ||||
| CRTAP | (CRTAP) Cartilage-assuciated protein | VII | АР | Тяжёлая ризомелия и белые склеры |
| P3H1 (LEPRE1) | (P3H1) Prolyl 3-hydroxylasel | VIII | АР | |
| PPIB | (PPlase B) Peptidyl-prolyl cis-trans isomerase B | IX | АР | Тяжёлые деформации конечностей и серые склеры |
| Нарушение созревания коллагена и присоединения шаперона | ||||
| SRPINH1 | (HSP47) Serpin Hl | X | АР | Тяжёлые скелетные деформации, синие склеры, несовершенный дентиногенез, аномалии кожи, паховая грыжа |
| FKBP10 | (FKBP65) 65kDa FK506 binding protein | XI | АР | Тяжесть скелетных деформаций варьирует от лёгких до тяжёлых форм, цвет склер — от нормального до серого; врождённые контрактуры |
| PLOD2 | (LH2) Lysyl Hydroxylase 2 | АР | Тяжесть скелетных деформаций варьирует от средних до тяжёлых форм; прогрессирующие контрактуры суставов | |
| BMP1 | (BMP1) Bone morphogenetic protein 1 | XII | АР | Тяжесть скелетных деформаций варьирует от лёгких до тяжёлых форм, пупочные грыжи |
| Нарушение дифференцировки и созревания остеобластов | ||||
| SP7 | (SP7) Transcription factor (osterix) | XIII | АР | Тяжёлые скелетные деформации, задержка прорезывания зубов, гипоплазия лица |
| TMEM38B | (TRIC-B) Trimeric intracellular caption channel type B | XIV | АР | Тяжёлые деформации конечностей, цвет склер варьирует от нормального до синего |
| WNT1 | (WNTI) Proto-oncogene Wnt-1 | XV | АР/АД | Тяжёлые нарушения развития скелета, белые склеры, неврологический дефицит |
| CREB3L1 | (OASIS) Old astrocyte-specificcally induced substance | XVI | АР | Тяжёлые деформации костей |
| SPARC | (SPARC) osteonectin | XVII | АР | Прогрессирующая хрупкость костей |
| MBTPS2 | XVIII | Х | Тяжесть скелетных деформаций варьирует от средних до тяжёлых форм; голубые склеры, сколиоз, деформация грудной клетки | |
Модифицированная классификация несовершенного остеогенеза [5]
| Типы несовершенного остеогенеза по новой классификации | Характеристика фенотипа | Тип несовершенного остеогенеза или заболевание |
| 1 | Лёгкое течение, без деформаций | I |
| 2 | Тяжёлое течение, перинатально летальное или летальное | II |
| 3 | От среднего до тяжёлого течения, с выраженными деформациями | III, VI, VIII, IX, X, синдром Брука 1-го типа |
| 4 | Средней тяжести, с широкой вариабельностью течения | IV, VII, XI, XII, XIII |
| 5 | Средней тяжести, включая костные патологии, приводящие к оссификации межкостных мембран | V, остеопороз-псевдоглиома, идиопатический ювенильный остеопороз, синдром Брука 1-го и 2-го типа |
Протокол внутреннего введения памидроната Montreal [7]
| Возраст | Дозировка | Частота введения |
| 0,5 мг/кг в день в течение 3 дней | Каждые 2 мес. | |
| 2–3 года | 0,75 мг/кг в день в течение 3 дней | Каждые 3 мес. |
| >3 лет | 1,0 мг/кг в день в течение 3 дней | Каждые 4 мес. |
Наследование
Если хотя бы у одного из родителей есть генетически обусловленная болезнь, то с вероятностью 50% это заболевание передастся ребёнку. У детей, которым не передался мутировавший ген, этого заболевания нет, и они не передают его дальше.
В подавляющем большинстве случаев наследованное нарушение вызвано новой мутацией. Это означает, что генетическая мутация происходит у человека впервые, а не передаётся по наследству. Следовательно, в семье, где есть ребёнок с новой мутацией, как правило, нет повышенного риска рождения другого ребёнка с этим заболеванием.
Если же два человека с НО решат завести малыша, то вероятность рождения ребёнка без врождённой хрупкости костей составит 25%.
В настоящее время для пациентов с НО существует возможность провести ЭКО.
II. Лечение и мониторинг
Общие положения о лечении
Лечение НО симптоматическое и зависит от тяжести течения. Цель лечения — снизить частоту переломов, помочь «хрупкому» человеку стать более подвижным и независимым, уменьшить болевой синдром. Кроме того, важно своевременно выявлять и контролировать внескелетные проявления и помогать пациенту избавиться от побочных эффектов лекарственной терапии.
Вести пациента с НО должна команда специалистов, состоящая из педиатра, эндокринолога, физического терапевта, травматолога-ортопеда, генетика, стоматолога, сурдолога, психолога, социального работника и т. д. Мультидисциплинарный подход — сочетание медикаментозного лечения, хирургии, ортопедии и реабилитации — значительно улучшает результат лечения.
На данный момент никакого лекарства от НО не существует. Кроме того, не существует и медикаментозной терапии, разработанной специально для лечения этого заболевания, — ни для детей, ни для взрослых. Лекарственная терапия, которая сейчас проводится, основана на препаратах, разработанных для лечения возрастного остеопороза или предотвращения потери костной массы у взрослых при онкологических заболеваниях и применяемых off-label — не по назначению в инструкции.
Укрепление костей в попытке уменьшить частоту и серьёзность переломов — ключевой вопрос лечения «хрупких» детей и взрослых. Именно поэтому лечение направлено на уменьшение влияния симптомов заболевания на организм человека и на предотвращение осложнений в будущем.
Часто требуется междисциплинарный план управления, который может включать в себя:
- Визиты к генетику (консультации и тестирование ДНК);
- Ортопедическое лечение переломов и деформаций;
- Проверку слуха;
- Обследование на наличие стоматологических проблем;
- Скрининг на наличие сердечно-сосудистых заболеваний (УЗИ сердца);
- Скрининг на респираторные проблемы (лёгочный тест);
- Физическую терапию;
- Эрготерапию с адаптацией приспособлений и окружающей среды;
- Психологическую помощь;
- Диетический совет — консультацию по питанию;
- Социальную помощь.
Для людей с НО очень важно полноценное питание, позволяющее поддерживать уровень уровень витамина D и кальция на необходимом уровне и полезное для здоровья костей. Следить за питанием важно ещё и потому, что люди с ограниченной подвижности склонны к набору веса, а лишний вес — это лишняя нагрузка на кости.
Следует уделить внимание профилактике остеопороза. Для людей с НО дополнительное воздействие остеопороза на скелет может иметь более серьёзные последствия, чем для остальных. Обратите внимание, что некоторые распространенные лекарства в списке возможных побочных эффектов имеют риск развития остеопороза.
Медикаментозное лечение
Несколько типов бисфосфонатов были использованы в качестве лечения для детей. По двум препаратам — памидроновой и золедроновой кислотам — есть международные протоколы .
Эффективность препаратов одинаковая, разница состоит в длительности введения:
- памидроновая кислота: три дня, три часа в день, раз в два-три месяца;
- золедроновая кислота: один день в течение часа, раз в три месяца.
В некоторых клиниках применяются и другие бисфосфонаты, например, препараты на основе ибандроновой кислоты. Лечение можно получить в государственных и частных клиниках.
Физическая реабилитация
Реабилитация — важнейший компонент комплексного подхода в лечении НО. Реабилитацию необходимо начинать с раннего возраста, чтобы помочь ребёнку адаптироваться к условиям окружающей среды и преодолеть страх переломов при освоении новых двигательных навыков.
Для людей с НО реабилитация — важнейший этап в восстановлении после травм, переломов, операций, а также в профилактике повторных переломов и деформаций. А если говорить о новорождённых, она направлена в первую очередь на обучение родителей безопасному обращению с «хрупким» малышом, которое необходимо для профилактики осложнений и для того, чтобы помочь ребёнку развивать двигательные навыки. Отмечено, что у пациентов с НО форма головы может быть треугольной и уплощённой, швы между костями черепа могут быть несросшимися и раскрытыми, а родничок — очень большим. Это изменится со временем. Часто встречаются и нарушения в тазобедренных суставах, которые обусловлены использованием мягких поверхностей для укладки ребёнка, сильно ограничивающих его в движении. Для профилактики нужно часто менять положение малыша. Когда ребёнок лежит на животе, распрямляются верхние конечности и шейный отдел позвоночника, а также растягиваются сгибатели бедра. Это помогает научиться переворачиваться и дальнейшем сидеть.
Самые первые шаги в лечебной физкультуре — контроль положения головы, укрепление мышц шеи, мышц, отвечающих за положение тела в пространстве и положение сидя с опорой на ноги. Физическая активность очень важна для «хрупких» детей, и начинать занятия нужно как можно раньше. К развитию двигательных навыков и активному плаванию в ванной можно приступать с рождения, в бассейн лучше тоже начать ходить, как только появится такая возможность. В старшем возрасте основное внимание уделяют увеличению мышечной силы, функциональному развитию и развитию самостоятельности.
Клиники, где лечат «хрупких»
Полный список российских клиник, в которых занимаются диагностикой и лечением несовершенного остеогенеза, представлен здесь .
III. Прогнозы
Из-за огромной индивидуальной изменчивости заболевания невозможно сделать общие заявления о том, какие методы лечения и реабилитации будут правильными в каждом конкретном случае. Могут быть высказаны лишь некоторые прогнозы, основанные на статистике для каждого типа НО, но нельзя дать никаких гарантий относительно количества переломов, роста «хрупкого» человека или его подвижности.
Даже у «хрупких» людей в пределах одной семьи различия могут быть весьма значительными, и в каждом конкретном случае рекомендуется обращаться за индивидуальной консультацией к специалисту.
Во многих случаях хрупкость костей уменьшается после полового созревания, но может снова увеличиться после сорока лет, поэтому физическая активность важна на протяжении всей жизни человека с НО.
Читайте также: