Глухота с ретинитом, слабоумием и прогрессирующим тетрапарезом
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Зрительный нерв является частью зрительного анализатора, обеспечивает передачу информации от сетчатки, воспринимающей изображение и преобразующей световую энергию в электрическую, к коре головного мозга, где формируется окончательное изображение.
Для атрофии характерным является снижение остроты зрения, очки и контактные линзы не корректируют зрение, наблюдается изменение в полях зрения, нарушение цветоощущения, начиная с зеленого и красного цветов, при осмотре - побледнение диска зрительного нерва.
Среди наследственной атрофии выделяют аутосомно-доминантную ( в сочетании с глухотой — синдром Ушера); и аутосомно-рецессивную (атрофия Бера, Йенсена и др.), а также митохондриальную (атрофия Лебера).
Синдром Ушера - это наследственная болезнь, характеризующаяся ухудшение слуха и прогрессирующим снижением зрения. Изменения зрения происходят из-за пигментного ретинита - дегенеративного заболевания сетчатки глаза, которое обычно проявляется в юности или на ранней стадии взросления. Иногда заболевание оказывает влияние и на вестибулярные функции человека. Проявления болезни могут меняться индивидуальным образом, а его прогресс может идти с различной скоростью.
Различают три различных формы синдрома Ушера. Первый тип связан с врожденной значительной тугоухостью и нарушением вестибулярных функций. Первые признаки пигментного ретинита - ночная слепота и ухудшение периферийного зрения - обычно наступают еще в юном возрасте.
При синдроме Ушера второго типа новорожденные дети отличаются средним или существенным нарушением слуха. Симптомы пигментного ретинита обычно начинают проявляться вскоре после периода отрочества. Ухудшение зрения происходит не так быстро, как в случае синдрома Ушера первого типа, а тугоухость обычно остается на стабильном уровне и не прогрессирует.
Более редкий синдром Ушера третьего типа - дети с этим заболеванием обычно рождаются с нормальным слухом или лишь с небольшим его нарушением. Прогрессирующее ухудшение слуха и зрения начинается лишь в возрасте полового созревания. Во многих случаях начинаются и вестибулярные нарушения.
Нарушения слуха при синдроме Ушера возникают из-за генетической мутации, оказывающей влияние на нервные клетки в ушной улитке, которая отвечает за слуховую часть внутреннего уха, воспринимающего и распознающего звуки. Тот же самый генетический дефект негативно влияет и на клетки фоторецепторов в сетчатке, что приводит к ухудшению зрения.
Синдром Ушера наследуется от родителей к их детям как аутосомно-рецессивная характеристика. При таком типе наследования каждый из родителей является носителем мутировавшего гена, но у них самих это никак не проявляется, поскольку каждый из них несет в себе только одну копию поврежденного гена. Ребенок имеет 25%-вероятность унаследовать мутировавший ген и заболеть.
Во всем мире синдром Ушера является одной из основных причин возникновения комбинированной слепоты и глухоты. Примерно 30% людей с пигментным ретинитом отмечают у себя частичную потерю слуха, и примерно половина из них имеет синдром Ушера. Чтобы оценить свое состояние и риск для других членов семьи или будущих детей заболеть синдромом Ушера, можно пройти молекулярно-генетическое тестирование на поиск мутаций в "горячих" участках гена ОРА1.
В состав гена ОРА1 входит 28 экзонов. Мутируют два экзона - 14 и 18. Мутации этих экзонов вызывают не только атрофию зрительного нерва с глухотой, но и миопатию и офтальмоплегию. Экзоны кодируют белок ОРА1.
Заболевание носит прогрессирующий характер и может привести к тяжелой потере зрения. Процент больных с потерей зрения составляет от 13 до 46 %. Важно вовремя диагностировать заболевание, так как у 25% больных оно протекает бессимптомно. Это затрудняет раннюю постановку диагноза.
Показания к назначению:
- подтверждение диагноза «атрофия зрительного нерва с глухотой»
- планирование беременности в семье, имеющей родственника с заболеванием атрофия зрительного нерва с глухотой
Метод исследования: секвенирование
Процедура взятия биоматериала оплачивается отдельно и зависит от типа материала:
Глухота и потеря слуха
Более 5% населения мира, или 430 миллионов человек, нуждаются в реабилитации для решения проблемы «инвалидизирующей» потери слуха (432 миллиона взрослых и 34 миллиона детей). По оценкам, к 2050 г. более 700 миллионов человек, или каждый десятый, будут иметь инвалидизирующую потерю слуха.
«Инвалидизирующей» называется потеря слуха в слышащем лучше ухе, превышающая 35 децибел (дБ). Почти 80% таких людей живет в странах с низким и средним уровнем дохода. Потеря слуха более широко распространена среди более возрастных людей: от этой проблемы страдают более 25% людей в возрасте старше 60 лет.
Потеря слуха и глухота
Человек, не способный слышать так же хорошо, как слышит человек с нормальным слухом – порог слышимости 20 дБ или ниже в обоих ушах, – страдает от потери слуха. Потеря слуха может быть легкой, умеренной, тяжелой или глубокой. Она может развиваться в одном или обоих ушах и затруднять слуховое восприятие разговорной речи или громких звуков.
Понятие «тугоухости» применяется по отношению к людям с потерей слуха, варьирующейся в пределах от легкой до тяжелой. Обычно тугоухие люди общаются с помощью разговорной речи и для улучшения слышимости могут пользоваться слуховыми аппаратами, кохлеарными имплантами и другими ассистивными устройствами, а также субтитрами.
«Глухие» люди в большинстве случаев страдают от глубокой потери слуха, то есть слышат очень плохо или не слышат вообще. Для общения такие люди часто используют язык жестов.
Причины потери слуха и глухоты
Хотя люди могут подвергаться влиянию перечисленных ниже факторов в разные периоды своей жизни, они наиболее восприимчивы к их воздействию в критические периоды жизни.
Предродовой период
- Генетические факторы: включают как врожденные, так и приобретенные причины потери слуха.
- Внутриутробные инфекции, такие как краснуха и цитомегаловирусная инфекция.
Перинатальный период
- Асфиксия при рождении (недостаток кислорода во время родов).
- Гипербилирубинемия (тяжелая форма желтухи в неонатальный период).
- Низкий вес при рождении.
- Другие перинатальные осложнения и их лечение.
Детский и подростковый возраст
- Хроническое воспаление среднего уха (хронический гнойный средний отит).
- Скопление жидкости в ухе (хронический негнойный средний отит).
- Менингит и другие инфекционные заболевания.
Взрослый и пожилой возраст
- Хронические заболевания.
- Курение.
- Отосклероз.
- Возрастная сенсоневральная дегенерация.
- Внезапная сенсоневральная потеря слуха.
Факторы, воздействующие на протяжении всей жизни
- Серная пробка (ушная сера, блокирующая слуховой проход).
- Травма уха или головы.
- Чрезмерный шум/громкие звуки.
- Ототоксичные лекарственные препараты.
- Ототоксичные химические вещества, связанные с работой.
- Недостаточность питания.
- Вирусные инфекции и другие болезни уха.
- Прогрессирующая наследственная потеря слуха с поздним началом.
К чему приводит потеря слуха, если не принимаются необходимые меры
Если не принимаются необходимые меры, потеря слуха может повлиять на множество аспектов жизни человека:
Общение и речь
Когнитивные функции
Образование и работа: в развивающихся странах дети, страдающие от потери слуха и глухоты, редко получают какое-либо образование. Среди взрослых людей, страдающих от потери слуха, отмечается гораздо более высокий уровень безработицы. По сравнению с общим работающим населением процентная доля работающих глухих людей, занимающихся менее квалифицированной работой, выше.
Социальная изоляция, одиночество и стигматизация
Социально-экономические последствия
Годы, прожитые с инвалидностью (YDL) и количество лет жизни, скорректированных с учетом инвалидности (DALY)
По оценкам ВОЗ, нерешенная проблема потери слуха ежегодно обходится миру в 980 млрд долл. США. Это включает расходы на здравоохранение (без учета стоимости слуховых аппаратов), расходы на помощь в процессе обучения, потери в результате утраты трудоспособности и социальные издержки. На страны с низким и средним уровнем дохода приходится 57% этих издержек.
Профилактика
Стратегии общественного здравоохранения и меры клинического вмешательства, принимаемые на протяжении всей жизни человека, позволяют избежать многих факторов, являющихся причинами потери слуха.
Профилактика потери слуха необходима на протяжении всей жизни – от пренатального и перинатального периодов до пожилого возраста. У детей потеря слуха почти в 60% случаев вызвана причинами, которые можно предотвратить, принимая меры общественного здравоохранения. Аналогичным образом, можно предотвратить наиболее распространенные причины потери слуха взрослыми, такие как воздействие громких звуков и ототоксичных лекарственных средств.
К числу эффективных мер, направленных на сокращение количества случаев потери слуха и принимаемых на разных стадиях жизни человека, относятся следующие:
- иммунизация;
- эффективная охрана здоровья матери и ребенка;
- генетическое консультирование;
- выявление и ведение наиболее распространенных болезней уха;
- программы защиты слуха от воздействия шума и химических веществ на производстве;
- стратегии пропаганды безопасного прослушивания с целью уменьшения воздействия громких звуков во время развлекательных мероприятий; и
- правильное применение лекарственных средств для предотвращения потери слуха под воздействием ототоксичных лекарственных средств.
Выявление и ведение
- Раннее выявление потери слуха и заболеваний уха имеет решающее значение для эффективного ведения пациентов.
- Для этого необходим систематический скрининг с целью выявления болезней ушей и связанной с ними потери слуха среди следующих категорий людей, подверженных наибольшему риску:
- новорожденные и грудные дети.
- дети дошкольного и школьного возраста.
- люди, подвергающиеся воздействию шума или химических веществ на работе.
- люди, принимающие ототоксичные лекарственные препараты.
- люди пожилого возраста.
- использование слуховых технологий, таких как слуховые аппараты, кохлеарные импланты и импланты среднего уха;
- использование языка жестов и других средств сенсорного замещения, таких как визуальное восприятие речи (чтение по губам), использование слепоглухими людьми метода тадома (прикладывание пальцев к губам и щекам говорящего), общение жестами; и
- реабилитационная терапия для улучшения навыков восприятия и развития коммуникативных и языковых способностей.
Деятельность ВОЗ
ВОЗ способствует внедрению комплексных и социально ориентированных систем охраны здоровья уха и слуха (IPC-EHHC).
Работая в этой области, ВОЗ руководствуется рекомендациями Всемирного доклада ВОЗ по проблемам слуха (2021 г.) и резолюцией о предупреждении глухоты и потери слуха Всемирной ассамблеи здравоохранения.
Глухота с ретинитом, слабоумием и прогрессирующим тетрапарезом
Глухота с ретинитом, слабоумием и прогрессирующим тетрапарезом
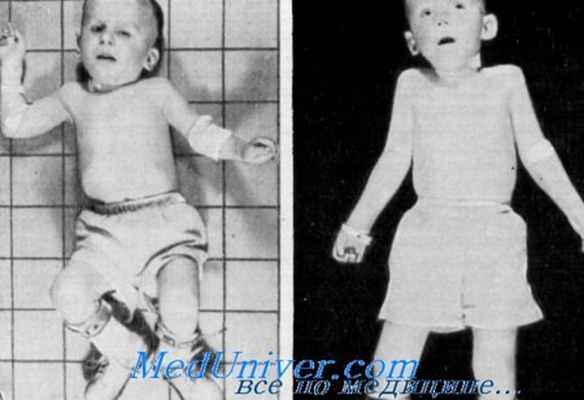
Пигментная дегенерация сетчатки, прогрессирующий тетрапарез, умственная отсталость и умеренно глубокая нсйросенсорная глухота у 2 мальчиков сибсов описаны Gordon, Capute и Konigsmark.
Клинические данные. Данные осмотра. Братья отличались низким ростом, атрофичной мускулатурой всех конечностей и тупым, невыразительным лицом. Окружность головы, длина тела и вес были ниже физиологических норм на 30%. Пальцы, особенно средние фаланги, были короткими. Отмечалась клинодактилия V пальцев. Наблюдались умеренные сгибательные контрактуры на ногах.
Орган зрения. Произведенное братьям в возрасте 3 и 4 лет офтальмологическое исследование обнаружило по всей сетчатке диффузное распространение крупных гранул пигмента, маленький и бледный диск зрительного нерва и суженный калибр сосудов (артериол) сетчатки. С возрастом все эти изменения становились более резкими.
Нервная система. Уровень IQ.s, установленный при психометрическом тестировании, был равен 34 и 44. Наиболее постоянным неврологическим симптомом болезни являлся прогрессирующий спастический паралич ног со сгибательными контрактурами бедер и коленей. Отмечались повышение сухожильных рефлексов и сгибательный подошвенный рефлекс. Несмотря на то что масса мышц была резко уменьшена, мышечная сила оставалась вполне хорошей. Больные ходили на широко расставленных ногах. Их походка была неустойчивой. Постепенно они ходили все с большим трудом, и в конце концов передвижение стало совсем невозможным.
Патологических движений и нарушения чувствительности не обнаружено. Выражение лица становилось все более тупым. Нарастали угнетение рвотного и глотательного рефлексов, а также нарушения смыкания век.
Орган слуха. Проведение аудиометрического исследования было затруднено из-за умственной отсталости мальчиков. Отологическое исследование патологии не выявило. ЭЭГ-аудиометрия показала умеренно глубокую пейросенсорную глухоту, охватывающую главным образом высокие частоты. Другие аудиометрические пробы не производились. Речь у обоих братьев не развивалась.
Вестибулярная система. Калорические вестибулярные пробы были нормальными.![глухота со слабоумием]()
Пигментный ретинит, прогрессирующий тетрапарез, слабоумие и нейро-сенсорная глухота. Два брата маленького роста с невыразительным лицом и низко посаженными деформированными ушами
Лабораторные данные. Рентгенограммы. На краниограмме обнаружены малые размеры и асимметрия черепа. Отмечались укорочение средних фаланг и гипоплазия V пястной кости.
Другие данные. На ЭЭГ у одного из мальчиков была обнаружена чрезмерно медленная активность, в то время как у его брата ЭЭГ была нормальная. Электроретинографическое исследование зарегистрировало у старшего мальчика почти нормальную реакцию на свет.Рутинные анализы мочи и крови, проба на электролиты и спинномозговая жидкость были нормальными.
Патология. Результаты гистопатологического исследования не представлены.Наследственность. Родители больших детей состояли в отдаленном родстве. У тетки по линии матери был спастический церебральный паралич, но без ретинопатии. Тип наследования более всего похож на аутосомно-рецессивный, но нельзя исключить возможность Х-сцепленного наследования.
Диагноз. При синдроме Ушера моторика и психическая сфера ингактны. Другие заболевания, включающие пигментный ретинит и глухоту, такие как синдром Кокейна, синдром Рефсума и синдром Альстрёма, имеют другие сопутствующие аномалии, отличные от спастической параплегии и умственной отсталости, наблюдающихся при этом заболевании.
Существует несколько синдромов, включающих умственную отсталость и нарушения походки. Синдром Ричарде — Раидля (Richards — Rundle) характеризуется глухотой и медленно прогрессирующей атаксией, но мышечная сила остается вполне хорошей в течение многих лет. Синдром Тройера (Тгоуег) включает спастическую параплегию и дистальиые мышечные атрофии, которые начинаются в детстве и медленно прогрессируют, пока в третьем — четвертом десятилетии жизни ходьба становится совершенно невозможной. Хотя у некоторых больных и выявляют умственную отсталость, у них не доказаны пигментный ретинит или атрофия зрительных нервов (Cross, McKusick).
В одной семье была описана спастическая параплегия с дегенерацией сетчатки в сочетании с нейросеисорной глухотой, но интеллект у больных был нормальным (Louis-Bar, Pirot, Mahloudji, Chuke)Лечение. Так как умственная отсталость у больных выражена резко, применение слуховых аппаратов ограничено.
Прогноз. Заболевание очень медленно прогрессирует, в результате развивается полная идиотия.Выводы. Характеристика этого синдрома включает: 1) вероятное аутосомно-рецессивное наследование, 2) прогрессирующий пигментный ретинит, 3) прогрессирующий тетрапарез, 4) выраженное прогрессирующее слабоумие, 5) умеренную нейросенсорную глухоту.
Синдромы, включающие пигментный ретинит и неврологические заболевания
Пигментная дегенерация сетчатки, прогрессирующий тетрапарез, умственная отсталость и умеренно глубокая нсйросенсорная глухота у 2 мальчиков сибсов описаны Gordon, Capute и Konigsmark.
Клинические данные. Данные осмотра. Братья отличались низким ростом, атрофичной мускулатурой всех конечностей и тупым, невыразительным лицом. Окружность головы, длина тела и вес были ниже физиологических норм на 30%. Пальцы, особенно средние фаланги, были короткими. Отмечалась клинодактилия V пальцев. Наблюдались умеренные сгибательные контрактуры на ногах.
Орган зрения. Произведенное братьям в возрасте 3 и 4 лет офтальмологическое исследование обнаружило по всей сетчатке диффузное распространение крупных гранул пигмента, маленький и бледный диск зрительного нерва и суженный калибр сосудов (артериол) сетчатки. С возрастом все эти изменения становились более резкими.
Нервная система. Уровень IQ.s, установленный при психометрическом тестировании, был равен 34 и 44. Наиболее постоянным неврологическим симптомом болезни являлся прогрессирующий спастический паралич ног со сгибательными контрактурами бедер и коленей. Отмечались повышение сухожильных рефлексов и сгибательный подошвенный рефлекс. Несмотря на то что масса мышц была резко уменьшена, мышечная сила оставалась вполне хорошей. Больные ходили на широко расставленных ногах. Их походка была неустойчивой. Постепенно они ходили все с большим трудом, и в конце концов передвижение стало совсем невозможным.
Патологических движений и нарушения чувствительности не обнаружено. Выражение лица становилось все более тупым. Нарастали угнетение рвотного и глотательного рефлексов, а также нарушения смыкания век.
Орган слуха. Проведение аудиометрического исследования было затруднено из-за умственной отсталости мальчиков. Отологическое исследование патологии не выявило. ЭЭГ-аудиометрия показала умеренно глубокую пейросенсорную глухоту, охватывающую главным образом высокие частоты. Другие аудиометрические пробы не производились. Речь у обоих братьев не развивалась.
Вестибулярная система. Калорические вестибулярные пробы были нормальными.Пигментный ретинит, прогрессирующий тетрапарез, слабоумие и нейро-сенсорная глухота. Два брата маленького роста с невыразительным лицом и низко посаженными деформированными ушами
Лабораторные данные. Рентгенограммы. На краниограмме обнаружены малые размеры и асимметрия черепа. Отмечались укорочение средних фаланг и гипоплазия V пястной кости.
Другие данные. На ЭЭГ у одного из мальчиков была обнаружена чрезмерно медленная активность, в то время как у его брата ЭЭГ была нормальная. Электроретинографическое исследование зарегистрировало у старшего мальчика почти нормальную реакцию на свет.Рутинные анализы мочи и крови, проба на электролиты и спинномозговая жидкость были нормальными.
Патология. Результаты гистопатологического исследования не представлены.Наследственность. Родители больших детей состояли в отдаленном родстве. У тетки по линии матери был спастический церебральный паралич, но без ретинопатии. Тип наследования более всего похож на аутосомно-рецессивный, но нельзя исключить возможность Х-сцепленного наследования.
Диагноз. При синдроме Ушера моторика и психическая сфера ингактны. Другие заболевания, включающие пигментный ретинит и глухоту, такие как синдром Кокейна, синдром Рефсума и синдром Альстрёма, имеют другие сопутствующие аномалии, отличные от спастической параплегии и умственной отсталости, наблюдающихся при этом заболевании.
Существует несколько синдромов, включающих умственную отсталость и нарушения походки. Синдром Ричарде — Раидля (Richards — Rundle) характеризуется глухотой и медленно прогрессирующей атаксией, но мышечная сила остается вполне хорошей в течение многих лет. Синдром Тройера (Тгоуег) включает спастическую параплегию и дистальиые мышечные атрофии, которые начинаются в детстве и медленно прогрессируют, пока в третьем — четвертом десятилетии жизни ходьба становится совершенно невозможной. Хотя у некоторых больных и выявляют умственную отсталость, у них не доказаны пигментный ретинит или атрофия зрительных нервов (Cross, McKusick).
В одной семье была описана спастическая параплегия с дегенерацией сетчатки в сочетании с нейросеисорной глухотой, но интеллект у больных был нормальным (Louis-Bar, Pirot, Mahloudji, Chuke)Лечение. Так как умственная отсталость у больных выражена резко, применение слуховых аппаратов ограничено.
Прогноз. Заболевание очень медленно прогрессирует, в результате развивается полная идиотия.Выводы. Характеристика этого синдрома включает: 1) вероятное аутосомно-рецессивное наследование, 2) прогрессирующий пигментный ретинит, 3) прогрессирующий тетрапарез, 4) выраженное прогрессирующее слабоумие, 5) умеренную нейросенсорную глухоту.
- Читать далее "Синдром Кокейна: карлик со старческим видом, умственной отсталостью и тугоухостью"
Система восстановления зрения.
Система М. С. Норбекова.
Занятия проводит преподаватель Центра академика М.С. Норбекова Екатерина Зенина.
Бесплатное ознакомительное занятие состоится 17 марта 2014 г. в 18:00.«Пусть во всех медицинских книгах вы прочтете, что ваша болезнь неизлечима, если что-то стоящее удерживает вас в этой жизни – вы обречены на успех».
Так утверждает человек, известный не только в нашей стране, но и далеко за ее пределами, Мирзакарим Санакулович Норбеков. В юности врачи признали его безнадежно больным. Но он сумел победить свой недуг. И теперь этот сильный человек показывает путь к здоровью другим.
Более 20 лет назад академик М.С. Норбеков создал оздоровительную систему, которая позволяет человеку любого возраста за десять занятий научиться самостоятельно наводить порядок в своем организме. В 1998 году по решению Международной ассоциации независимых медицинских экспертов она признана самой эффективной среди других альтернативных оздоровительных систем.
Система ускоренного обучения саморегуляции организма предлагает десять путей выхода из нездоровья. Это и суставная гимнастика, и гимнастика воли, и гимнастика воображения, и тренировка эмоций и многое другое. Но основа – в том, что человек из неудачника превращается в Победителя.
Формула Норбекова: волевое принуждение – мышечный корсет – настроение – вера – результат! Хотите – верьте, хотите – проверьте! Не забудьте, что проверять надо не менее сорока дней и обязательно создавать при этом внутреннее состояние того, что вы Победитель!
Но для получения результата надо потрудиться. Работа над собой требует преодоления лени, поэтому в аудитории только те, кто к этому готов. Это единственное условие выздоровления. Иначе любые теоретические знания бесполезны. И Наставник и Ученик, каждый – должен пройти свою половину пути, суть которого – настрой на добро и жизнь.
За 10 дней Вы сможете: восстановить зрение и слух; избавиться от боли в позвоночнике; нормализовать давление; укрепить середечно-сосудистую и нервную системы; избавиться от бессоницы; укрепить эндокринную и имунную системы; решить гинекологически-урологические проблемы; очистить и омолодить свой организм; устранить дефекты кожи; регулировать дыхание и питание; забыть об апатии и депрессии; наладить семейные отношения; создать личную модель успеха; раскрыть свои внутренние возможности; узнать о секретах древневосточной медицины. Мы учимся исцелять себя и поддерживать свое здоровье естественными методами. Мы освоим навыки: формирования внутренней уверенности; избавления от вредных привычек; создания положительных привычек; развития навыков по Вашему выбору; постановки и достижения целей.
Учебно-оздоровительный курс по методике М.С.Норбекова направлен на обучение наших слушателей базовым возможностям управления своим организмом, вплоть до клеточного уровня.
Результатом практического освоения материалов этого курса является:
- фактическое восстановление физического здоровья;
- повышение сопротивляемости иммунной и прочих жизненно важных систем к неблагоприятным воздействиям окружающей среды;
- ускоренная регенерация тканей и, как следствие, омоложение человека;
- качественное улучшение ежедневного самочувствия, повышение общего тонуса и работоспособности.
Многие вначале сомневаются, что можно быть здоровым при помощи всего лишь каких то физических и мысленных упражнений. Не глотая пачками таблетки и пилюли получать удовольствие от здоровой полноценной жизни. Конечно, Вы можете продолжать дальше жить, как привыкли: не изменяя своим стереотипам, не веря в собственные силы и не используя возможности, которые Природа заложила в нас с детства.
Для тех, кто желает изменять свою жизнь к лучшему, мы предлагаем начать со здоровья, используя проверенную эффективную методику с маленькими восточными хитростями, адаптированными к европейскому восприятию. Она позволит Вам легко и быстро получить ощутимые результаты по улучшению своего здоровья и самочувствия уже в течение первых дней занятий.
СКИДКА 20% на весь курс предоставляется студентам, пенсионерам, инвалидам, а также двум или более представителям одной семьи.
План: Понятия о липидозов. Классификация. Литературы. Липидозы
Липидозы.
Липидозы (lipidoses; липид [ы] + -ōsis) — группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Л. относится к болезням накопления, которые обусловлены отложением аномально больших количеств нерасщепленных продуктов жирового обмена в различных органах и тканях, что приводит к значительному нарушению их функции. В основе Л. лежит полная или частичная недостаточность лизосомальных ферментов, участвующих в обмене липидов и обусловленная наследственным дефектом соответствующего гена. Большинство Л. наследуется по аутосомно-рецессивному типу, исключение составляет болезнь Фабри, которая наследуется по Х-сцепленному, рецессивному типу.
Липидозы включают липопротеинемии, связанные с нарушением обмена липопротеидов (см. Дислипопротеинемии), и гликолипидозы, обусловленные нарушением обмена гликолипидов.
В организме человека из гликолипидов наиболее распространены сфингогликолипиды, поэтому заболевания, связанные с нарушением их обмена (сфинголипидозы), составляют основную часть гликолипидозов. Для обозначения гликолипидозов используют термин «липидозы».Классификация.
К липидозам относятся ганглиозидозы — GM1-ганглиозидоз (тип I, II, III), GM2-ганглиозидоз (тип I, II, ювенильный, хронический тип); болезнь Ниманна — Пика; болезнь Гоше; болезнь Фабри; липогранулематоз Фарбера; болезнь Вулмена; болезнь I-клеток; псевдогурлевскую полидистрофию; метахроматическую лейкодистрофию; болезнь Краббе.Амавротическая идиотия (цереброретинальная дегенерация) - группа наследственных заболеваний, которые характеризуются прогрессирующим снижением зрения и развитием деменции в сочетании со спастическими параличами и другими неврологическими симптомами.
Чаще других форм встречается инфантильная форма Тея-Сакса - наиболее известная болезнь из группы наследственных липидозов. Наследуется по аутосомно-рецессивному типу. В отдельных популяционных группах населения, связанных с изолятами, частота заболевания равна 1:5000 новорожденных. В патогенезе ведущая роль отводится дефициту фермента бета-гексозаминидазы А в плазме крови, мозге и внутренних органах, что приводит к накоплению в их клетках ганглиозида Gm2. Клиническая картина заболевания проявляется в возрасте 4-6 месяцев. Типичными синдромами болезни являются повышение мышечного тонуса, переходящее в децеребрационную ригидность, и задержка психического развития до степени идиотии. Рано обнаруживается снижение зрения. На глазном дне в макулярной области выявляется пятно вишневого цвета ("вишневая косточка") на фоне атрофии зрительного нерва. Больные погибают в возрасте 2-3 лет.
Сходная клиническая картина (нарастающая деменция, атрофия зрительных нервов или пигментный ретинит, спастические парезы и параличи в сочетании с другими симптомами поражения нервной системы) характерна и для более редких вариантов амавротической идиотии - врожденной формы Нормана-Вуда, ювенильной формы Баттена-Шпильмейера-Фогта, позднедетской формы Билыповского- Янского, поздней формы Куфса. Клинический диагноз амавротической идиотии основывается на данных клинической картины заболевания, характерных изменениях глазного дна и определении активности специфического фермента.
Болезнь Ниманна-Пика (сфингомиелиноз) - наследственное нарушение обмена сфингомиелина, при котором происходит накопление сфингомиелина в мозге, печени, селезенке, ретикулоэндотелиальной системе. Тип наследования - аутосомно-рецессивный. Биохимический дефект обусловлен дефицитом фермента, катализирующего гидролиз фосфохолина из сфингомиелина. При этом происходит накопление и отложение сфингомиелина и, параллельно, холестерина в клетках ретикулоэндотелиальной системы и в мозге. Различают раннюю тяжелую форму заболевания, при которой у детей с первых дней жизни наблюдаются отказ от пищи, периодическая рвота, обезвоживание организма, увеличиваются печень и селезенка. Наряду с этим происходит задержка психического развития, развиваются спастические парезы, нарушается координация движений, возникают глухота и слепота. Слабоумие прогрессирует, развивается децеребрационная ригидность. На глазном дне - атрофия сосков зрительных нервов, в 20-30% случаев - вишневое пятно. Гибель наступает через 2-3 года от начала заболевания. При поздней хронической форме заболевания, возникающей в подростковом возрасте или у взрослых, болезнь течет длительно, нервная система обычно не поражается или вовлекается в патологический процесс в позднем периоде.
Диагноз болезни Ниманна-Пика основывается на данных клинической картины, исследовании глазного дна, определении активности специфического фермента, обнаружении клеток Ниманна-Пика в стернальном пунктате или в пунктатах селезенки.
Болезнь Гоше (глюкоцереброзидоз) - наследственно обусловленное нарушение обмена глюкоцереброзидов, при котором они накапливаются в клетках ретикулоэндотелиальной системы. Тип наследования - аутосомно-рецессивный. Обнаружен дефицит фермента бета-глюкоцереброзидазы, катализирующего отщепление глюкозы от глюкоцереброзида. Это приводит к накоплению цереброзидов и отложению их в клетках ретикулоэндотелиальной системы. Заболевание характеризуется развитием спленомегалического синдрома, анемии и поражения костной системы. Неврологическая симптоматика заболевания вариабельна. Детская тяжелая форма заболевания сопровождается задержкой общего и психического развития. Эпилептические припадки являются одним из постоянных симптомов заболевания. Развивается слепота (пигментная дегенерация сетчатки), глазодвигательные нарушения, бульбарные расстройства. Смерть наступает через 1,5-2 года. У взрослых заболевание отличается хроническим течением и протекает более доброкачественно. Нервная система в патологический процесс вовлекается редко. Клиническая картина складывается из гепатоспленомегалии, анемии, геморрагического синдрома, поражения трубчатых костей. Остеодистрофии могут являться причиной спонтанных переломов конечностей. Смерть наступает в связи с ослаблением иммунологической реактивности.
Диагноз основывается на клинической картине заболевания, подтверждается исследованием активности глюкоцереброзидазы в лейкоцитах и кожных фибробластах. В пунктатах костного мозга или селезенки обнаруживаются клетки Гоше.
Болезнь Рефсума (полиневритоподобная гемералопическая гередоатаксия) - редкое заболевание из группы липидозов. Наследуется по аутосомно-рецессивному типу. Заболевание обусловлено блокадой нормального альфа-окисления фитановой кислоты (присутствующей в пище как компонент растительных и животных жиров) в альфа-гидроксифитановую, что приводит к накоплению фитановой кислоты в крови и тканях. Неврологические нарушения, по-видимому, прямо связаны с накоплением кислоты в центральной и периферической нервной системе. Клинические проявления возникают, как правило, между 10 и 30 годами. Основным синдромом является сенсо-моторная полиневропатия. Характерны пигментная дегенерация сетчатки, мозжечковая атаксия, глухота, аносмия, зрачковые нарушения, деформации скелета, кардиомиопатия. Течение заболевания медленно прогрессирующее. Диагностика базируется на типичных клинических проявлениях, семейном анамнезе, биохимических показателях - высоком содержании фитановой кислоты в сыворотке крови и повышенном выделении с мочой жирных кислот.
Лейкодистрофии (прогрессирующий распад белого вещества) - редкие заболевания, представляющие большие трудности для прижизненной диагностики. Наследуются преимущественно по аутосомно-рецессивному типу, чаще начинается в дошкольном возрасте, преимущественно болеют мальчики. Для этой группы заболеваний характерно поражение белого вещества (демиелинизация) головного и спинного мозга, типична симметричность поражения.
Клиническая картина лейкодистрофии весьма полиморфна. Наиболее характерны нарастающие пирамидные, экстрапирамидные, мозжечковые расстройства, нарушения слуха И зрения (атрофия зрительных нервов), эпилептические припадки, прогрессирующая деменция.
Наиболее известны следующие формы лейкодистрофии: метахроматическая лейкодистрофия Шольца-Гринфилда, глобоидно-клеточная лейкодистрофия Краббе-Бенеке, суданофильная лейкодистрофия Пелициуса-Мерцбахера (см.).
Дифференциальный диагноз проводится с опухолями головного мозга и лейкоэнцефаяитами. Нарастающая внутричерепная гипергензия, очерченный очаговый синдром, результаты компьютерной томографии головного мозга свидетельствуют об опухоли головного мозга. Выявление инфекционного фона, аутоиммунный характер заболевания, отсутствие наследственной отягощенности говорят в пользу лейкоэнцефалита. Прогредиентность течения заболевания, системность патологического процесса, семейный анамнез, активность ферментов и уровень липидов в крови и спинномозговой жидкости помогают констатировать наличие лейкодистрофии.
Лечение. Этиотропная терапия липидозов не разработана, патогенетическая - весьма ограничена. Используют средства, направленные на нормализацию липидного обмена (плазмаферез, введение тканевых экстрактов, витаминов, ненасыщенных жирных кислот). Ремиелинизаторы (ретаболил, келтикан), ноотропные препараты (церебролизин, пирацетам), противосудорожные средства. При болезни Рефсума - общеукрепляющая терапия, антихолинэстеразные препараты, массаж, лечебная физкультура. Целесообразно исключить из рациона питания продукты, содержащие фитановую кислоту и хлорофилл.
Болезнь Краббе (глобоидно-клеточная лейкодистрофия) — быстро прогрессирующее демиелинизирующее дегенеративное заболевание ц.н.с. Тип наследования — аутосомно-рецессивный. В основе болезни лежит снижение активности фермента галактозилцерамид-b-галактозидазы, который в норме расщепляет галактоцереброзид до церамида и галактозы. В головном мозге, печени, селезенке, почках, лейкоцитах, фибробластах накапливаются галактоцереброзид и его производное психозин. Количество последнего повышается в 10—100 раз, что оказывает токсическое воздействие на олигодендроглиальные клетки, формирующие миелиновую оболочку. В зонах демиелинизации вокруг мелких кровеносных сосудов белое вещество содержит большое количество глобоидных гистиоцитов (макрофагов). Уменьшение олигодендроглиальных клеток сопровождается глиозом. Периферические нервы подвергаются аксональной дегенерации с накоплением пенистых гистиоцитов.
Как правило, первые клинические симптомы появляются на четвертом месяце жизни. Отмечаются повышенная возбудимость и мышечная гипертония. Верхние и нижние конечности у больных детей находятся в разогнутом состоянии, кулаки сжаты. Вскоре становится заметной задержка психомоторного развития. Позднее развиваются миоклонические судороги, генерализованная двигательная реакция на слуховые раздражители, спастический тетрапарез, повышение глубоких сухожильных рефлексов, при исследовании глазного дна выявляют атрофию зрительных нервов. Проявления периферической нейропатии встречаются лишь в отдельных случаях. В последующем развиваются характерные глубокие умственные расстройства, вялый тетрапарез, снижаются сухожильные рефлексы, мышечная гипертония сменяется гипотонией. Больные умирают в возрасте от 7 месяцев до 3 лет.
Диагноз основан на клинической картине, повышении содержания белка в цереброспинальной жидкости, выявлении снижения активности галактозилцерамид-b-галактозидазы. Дифференциальный диагноз проводят с другими формами лейкодистрофии на основании биохимического анализа. Специфическое лечение не разработано. Возможна пренатальная диагностика болезни Краббе путем исследования в культивируемых амниотических клетках активности галактозилцерамид-b-галактозидазыПричины липидозов.
Причиной липидозов являются наследственно обусловленные дефекты ферментных систем лизосом. В результате страдает нормальный жировой обмен, в клетках накапливается большое количество нерасщепленных сложных липидов в виде хиломикронов - частиц нейтрального жира размером около 1 мкм. Заболевания этой группы всегда носят системный характер и протекают с поражением центральной нервной системы.
Кожные проявления наблюдаются в той или иной степени практически при всех липидозах, но чаще всего в практике дерматокосметолога встречается болезнь Фабри (диффузная ангиокератома туловища).
Исход.
Исход: часто – обратимые изменения (при отсутствии серьезных разрушений клет. Структур.Читайте также: