Краниодиафизарная дисплазия: клиника, диагностика, наследственность
Добавил пользователь Дмитрий К. Обновлено: 28.01.2026
Рецессивная краниометафизарная дисплазия
В клиническом и морфологическом отношении это заболевание более тяжелое, чем доминантная краниометафизарная дисплазия. В течение нескольких первых лет жизни отмечается изменение черт лица, выражены гипертелоризм и широкое переносье. Постоянным симптомом является поражение черепно-мозговых нервов.
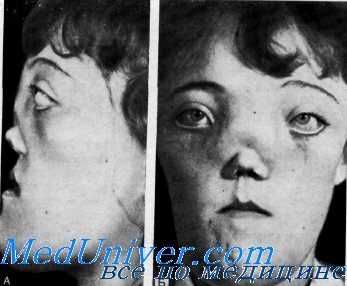
Клинические данные. Данные осмотра. Черты лица уродливы. Обращают внимание выступающие вперед переносье и иараназальные области и/или резко выраженный нижнечелюстной прогнатизм. Обычно наблюдается полное закрытие носовых ходов, что выражается в постоянно открытом рте.
Интересно отметить, что у женщин сибсов, описанных Millard с соавт. и Ross и Altman, наблюдалось поражение нижней челюсти, тогда как у братьев этого не было. И у тех, и у других отмечалось резкое смещение костей в области носа.
Костная система. Костные аномалии описаны выше — см. «Данные осмотра», и ниже — см. «Лабораторные данные».
Орган зрения. Постоянной чертой болезни является гипертелоризм. Слепота в 7-месячном возрасте вследствие атрофии зрительных нервов была описана Jackson с соавт. в их наблюдении № 4. У больного, о котором сообщил Graf, развилась умеренная потеря зрения, резче выраженная с одной стороны. Неклассифицированная прогрессирующая потеря зрения была отмечена Sommer. Атрофия зрительных нервов не обнаружена.
Нервная система. У больного, описанного Graf, развился односторонний парез лицевого нерва. В наблюдении № 5, представленном Jackson с соавт., описанного также Thorna и Goldman, в возрасте 21 года развился паралич лицевого нерва. Умственная отсталость была отмечена в наблюдении № 4, описанном Jackson с соавт.
Орган слуха. У многих больных отмечалась глухота. Резко выраженная потеря слуха обнаружена Jackson с соавт. в наблюдении № 5, а также Sommer, у его больного в возрасте от 10 до 15 лет. У больного, описанного Graf, глухота была выявлена в 6-летнем возрасте. Аудиометрия показала потерю слуха, равную 90 дБ по воздушной проводимости и от 20 до 50 дБ по костной проводимости. Полученные данные позволили предположить, что преобладает проводящая глухота, развивавшаяся, возможно, в связи с анкилозом основания стремени.
Вестибулярная система. Результаты вестибулярных проб не описаны.
Лабораторные данные. Рентгенограммы. Отмечались многочиленные аномалии черепа и липа, включающие гиперостоз свода черепа, расширение коркового слоя черепа, склероз основания черепа, гниертелоризм, отсутствие параназальных синусов, заполнение костью отверстий черепно-мозговых нервов, задержку прорезывания постоянных зубов и инжнечелюстной прогнатизм (Sommer, Lievre, Fischgold).
Co стороны длинных костей было обнаружено расширение метафизов, резче всего в диетальных отделах бедра, похожее на то, что наблюдалось при доминантной форме болезни. Отмечался склероз. Степень его выраженности была между тем, что наблюдается при доминантной краниометафизарной дисплазии, и тем, что отмечается при краниодиафизарной дисплазии.
Другие данные. Лабораторные исследования не выявили патологии со стороны крови, сыворотки крови, мочи и спинномозговой жидкости,
Наследственность. Заболевание наследуется по аутосомно-рецессивному типу. (Зольные сибсы со здоровыми родителями были описаны Lehmann и Millard с сотр.. Кровное родство родителей было доказано в наблюдениях, описанных Lievre н Fischgold, а также Graf.
Диагноз. Рентгенологические находки похожи на те, что наблюдаются при доминантной форме болезни. Краниометафизарную дисплазию доминантную или рецессивную легко спутать с метафизарной дисплазией (болезнью Пиле). Последнее заболевание наследуется по аутосомно-рецессивному типу и сочетается с более грубыми общими изменениями костей, однако кости лица и черепа не поражаются (Gorlinet al.).
Лечение. Костную дисплазию можно лечить хирургическими методами (Millard et al.). Для улучшения слуха можно использовать слуховые аппараты. С успехом можно заменить пораженные слуховые косточки протезом.
Прогноз. Костные изменения медленно прогрессируют. В результате разрастания костей в отверстиях черепа могут развиться параличи черепно-мозговых нервов.
Выводы. Главными чертами этого заболевания являются: 1) аутосомно-рецессивный тип наследования; 2) характерное «львиное» лицо; 3) типичные рентгенологические изменения, включающие гиперостоз свода и основания черепа, расширение метафнзов длинных костей с деформацией типа Erlenmeyer-flask; 4) параличи черепно-мозговых нервов; 5) смешанная, но преимущественно проводящая глухота.
Краниодиафизарная дисплазия: клиника, диагностика, наследственность
Лобно-метафизарная или фронто-метафизарная дисплазия
Gorlin и Cohen выделили лобно-метафпзарную дисплазию из многих других краниотубулярных дисплазий. Это заболевание проявляется выбухающими костными надглазничными утолщениями, проводящей глухотой и различными изменениями скелета. Похожие случаи были описаны Lischi, Walker, Holt с соавт., Danks с соавт., Jarvis и Jenkins, Weiss и Reynolds.
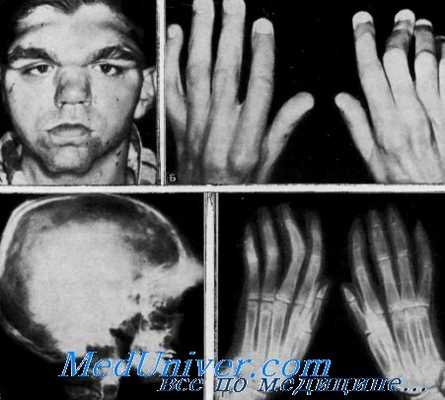
Клинические данные. Данные осмотра. Выраженные надглазничные бугры, широкий корень носа и маленький острый подбородок придают больным странный внешний вид. Утолщение надглазничных дуг становится очевидным к пубертатному возрасту (Danks et al.).
Костно-мышечная система. Отмечаются атрофии мышц рук п ног, особенно гипотенара и межкостных мышц кистей. Полное дорсальное сгибание кисти и разгибание локтей невозможны, пронация и супинация также чрезвычайно органичены. Прогрессируют сгибательные контрактуры пальцев и ульнарное отклонение кисти. Движения пальцев ограничены, особенно в пястно-фаланговых суставах. На ногах, кроме того, пальцы имеют молоткообразную форму.
Нервная система. Walker отметил головную боль и снижение чувствительности в областях, снабжаемых тройничным нервом. Jarvis и Jenkins сообщили о глубокой умственной отсталости у их больных.
Другие данные. У одного больного наблюдался маленький половой член и крииторхизм (Gorlin, Cohen). Блок одной из ножек пучка Гиса и гипертрофия правого желудочка были отмечены Danks с сотр.
Наблюдались отсутствие постоянных зубов при сохранности молочных зубов, а также расщепление язычка (Lischi, Gorlin, Cohen, Danks et al.).
Орган слуха. Gorlin и Cohen описали умеренную проводящую глухоту у 19-летнего больного. Walker сообщил о дефиците слуха и приступообразном шуме в ушах, выявившихся у наблюдаемого им больного к 25-летпему возрасту. Глухота не была охарактеризована. Holt с сотр. и Arenberg с соавт. обнаружили симметричную прогрессирующую смешанную глухоту, выявившуюся впервые к пубертатному возрасту. При тимпанотомии была обнаружена фиксация молоточка и наковальни.
Вестибулярная система. Результаты обследования вестибулярной системы не опубликованы.
Лабораторные данные. Рентгенограммы. При рентгенологическом исследовании были обнаружены толстые, валикоиодобиые лобные бугры, отсутствие лобных синусов, дефекты надорбитальных щелей и дуг верхнего края верхнечелюстных синусов в форме «купола мечети», а также зазубрины вдоль нижнего края нижней челюсти до углов, выраженную гипоплазию угла и мыщелка нижней челюсти (Gorlin, Cohen, Holt et al.).
Большое отверстие черепа было резко расширено. Отмечались множественные аномалии шейных позвонков, такие, как расположенный далеко кпереди зубовидный отросток, отсутствие задней дуги атланта, синостоз С2 и С3, частичное смещение С3 и С4. На рентгенограммах длинных костей обнаружены повышенная плотность диафизов и недостаточно сформированные метафизы, что создает деформацию типа зрленмейеровских колб (Erlenmeyer-flask). Отмечались выпуклые подвздошные кости, расширенные и удлиненные средние фаланги пальцев, тонкие, слабые ребра. При обследовании позвоночника выявлено увеличение расстояния между ножками дуг в поясничном отделе (Danks et al.). Ребра и позвонки были неправильно контурированы (Holt et al.).
Другие данные. Danks с сотр. обнаружили метахромазию в культе фибробластов.
Наследственность. Болезнь может быть генетически неоднородной. Weiss и Reynolds описали заболевание у матери и сына, предположив аутосомно-доминантное наследование. Jarvis и Jenkins сообщили о заболевании у мальчиков полусибсов, имевших одну мать, и установили Х-сцепленное наследование. Несмотря на сходство костных аномалий, больные были глубоко умственно отсталыми, что не было обнаружено пи в одном другом случае.
Диагноз. Необходимо исключить другие краниотубулярные дисплазии, такие, как краниометафизарная дисплазия и краниодиафизарная дисплазия. Эти заболевания подробно обсуждены Gorlin с соавт..
Толстые, валикоподобные лобные бугры наблюдаются в конечной стадии метафизарного дизостоза Янсена (Jansen), заболевания костной системы, полностью отличающегося со всех других сторон, от лобно-метафизарной дисплазии (de Haas et al.).
Лечение. Для улучшения слуха можно использовать слуховые аппараты.
Выводы. Характеристика этого синдрома включает:
1) аутосомно-доминантное или Х-сцепленное рецессивное наследование;
2) характерное лицо, отличающееся выбухающими надглазничными буграми и маленьким подбородком;
3) атрофии мышц рук п ног со сгибательными деформациями суставов;
4) характерные изменения скелета;
5) смешанную, чаще проводящую глухоту.
Gorlin, Spranger и Koszalka применили термин «краниодиафизарная дисплазия» для обозначения очень тяжелого заболевания костной системы, характеризующегося массивным генерализованным гиперостозом и склерозом, охватывающим главным образом кости черепа и лица (de Souza, Halliday, Stransky et al., Macpherson). Больной, описанный Gemmell, имел, вероятно, легкую форму болезни.
Клинические данные. Данные осмотра. Лицо и череп резко утолщены, расширены и деформированы. В течение нескольких первых лет или даже нескольких первых месяцев жизни становится очевидной закупорка носа и наблюдаются повторные респираторные инфекции. Выражены утолщение костей носа, гипертелоризм и уплощение носа, впоследствии развивается общее резко выраженное несмыкание зубов. Двусторонний стеноз хоан выявляется в течение первых нескольких лет жизни, и больные вынуждены для дыхания постоянно держать открытым рот.
Рост задержан. Больные обычно умирают в молодом возрасте. Костная система. Костные изменения были описаны выше (см. «Данные осмотра») н будут описаны ниже.
Орган зрения. У всех больных имеются резко выраженный гипертелоризм, закупорка слезного капала в результате разрастания костей, а также снижение остроты зрения или слепота вследствие атрофии зрительных нервов (de Souza, Gemmell, Halliday, Joseph et al.).
Нервная система. Все этапы развития задержаны. В результате разрастания костной ткани возникает сдавлеиие черепно-мозговых нервов. Этот безжалостный процесс приводит к возникновению головной боли, развитию прогрессирующего слабоумия и судорожных припадков (Gemmell, Halliday, Stransky et al.). Заметно повышается внутричерепное давление (Macpherson). Возможно, вследствие повреждения гипофиза отмечается задержка полового развития (de Souza, Stransky et al.).
Орган слуха. Глухота обнаружена во всех случаях. У некоторых больных потеря слуха была врожденной (Stransky et al.). Gemmell охарактеризовал глухоту как смешанную, Halliday как нейросенсорную.
Вестибулярная система. Результаты исследования вестибулярной системы не представлены.
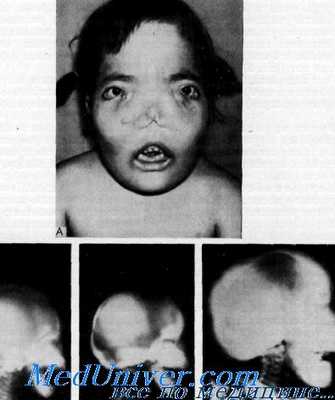
Лабораторные данные. Рентгенологически кости черепа, лица и нижняя челюсть резко склерозированы и гиперостозированы. Параназальные синусы и клетки сосцевидных отростков не развиты. Отмечаются умеренное утолщение и выраженный склероз ребер и ключиц. На длинных трубчатых костях не выражены выпуклости метафизов, виден эндостоз диафизов. По своей форме кости скорее всего напоминают «ночную полицейскую дубинку». Короткие трубчатые кости рук и ног, особенно первые пястные и плюсневые кости, цилиндрической формы. Костный возраст может быть задержанным.
Joseph с соавт., Stransky с соавт. и Macpherson обнаружили в сыворотке крови повышенный уровень алкалинфосфатазы, но нормальный уровень кальция и фосфора.
Наследственность. Все приведенные выше наблюдения, за исключением сибсов (мальчика и девочки), описанных de Souze, были спорадическими. Родители были здоровы. Кровное родство между родителями отмечено Halliday. Заболеванием поражаются и мальчики, и девочки. Так, что наиболее вероятен аутосомно-рецессивный тип наследования.
Диагноз. При краниометафизарной дисплазии, при обеих формах: доминантной и рецессивной, наблюдаются гипертелоризм, параназальные костные разрастания и сужение черепных отверстий. При обоих заболеваниях отмечаются легкие изменения со стороны метафизов длинных костей, особенно бедренной кости, что явно отличает эти две формы от описываемого здесь заболевания.
Лечение. Лечение невозможно.
Прогноз. Прогноз неблагоприятный. Заболевание безжалостно прогрессирует и ведет к глубокой умственной отсталости, а также к нарушению физического развития, слепоте и глухоте. Очень часто больные рано умирают.
Выводы. Характеристика этого заболевания включает: 1) аутосомно-рецессивное наследование; 2) массивное увеличение и склероз костей лица, черепа, ребер и ключиц; 3) цилиндризацию длинных костей и эндостозы диафизов; 4) костные разрастания в отверстиях черепа, в результате чего развиваются слепота и глухота; 5) повышение уровня алкалинфосфатазы в сыворотке крови; 6) смешанную глухоту.
Краниометафизарная дисплазия
Краниометафизарная дисплазия – наследственное состояние из группы остеохондродисплазий, характеризующееся аномалиями развития черепа и метафизов костей конечностей. Симптомами данного заболевания являются гипертелоризм, пороки развития лица, нередко уродующие больного, аномальное формирование носовых ходов с нарушением их проходимости, иногда головные боли и искривления конечностей. Диагностика краниометафизарной дисплазии осуществляется на основании изучения настоящего статуса пациента, рентгенологических данных и результатов молекулярно-генетических исследований. Специфического лечения данной патологии не существует, используют симптоматическую терапию, в некоторых случаях проводят хирургические вмешательства для облегчения дыхания пациента и улучшения его внешнего вида.
Общие сведения
Краниометафизарная дисплазия – группа наследственных заболеваний, которые приводят к порокам развития костей лицевого и мозгового отделов черепа различной степени выраженности в сочетании с аномалией метафизов длинных трубчатых костей и другими нарушениями. Ранее это состояние относили в одну группу с болезнью Пайла (множественная метафизарная дисплазия, краниометафизарная дисплазия Пайла), однако в настоящее время его выделяют в отдельную нозологическую единицу. Это связано с тем, что при данном заболевании превалирующими являются именно аномалии строения черепа (деформации, гиперостозы и склерозы костей), тогда как патологии иных отделов скелета выражены слабо. Существуют две основные формы краниометафизарной дисплазии, отличающиеся между собой механизмом наследования (аутосомно-доминантный и аутосомно-рецессивный типы), клиническими проявлениями и выраженностью нарушений. За счет наследования посредством аутосом патология с равной долей вероятности поражает как мужчин, так и женщин. Встречаемость краниометафизарной дисплазии точно не установлена, доминантный тип регистрируется во много раз чаще рецессивного.
Причины краниометафизарной дисплазии
Основной причиной более частой, но более легкой аутосомно-доминантной формы краниометафизарной дисплазии становятся мутации в гене ANKH, располагающемся на 5-й хромосоме. Ген кодирует белок, который является мембранным переносчиком пирофосфата, участвующего в угнетении процессов обызвествления костной ткани и ее резорбции. В результате генетического дефекта у белка-переносчика изменяется структура, и он становится неспособным полноценно выполнять свои функции, что приводит к краниометафизарной дисплазии. На клеточном уровне это проявляется аномальным изменением активности остеокластов, развитием склероза и гиперостоза главным образом костей черепа. Также может нарушаться конфигурация основных отверстий основания черепа, что становится причиной компрессии некоторых сосудов и нервов и во многом определяет остальные симптомы краниометафизарной дисплазии (нарушения слуха, головные боли, поражения тройничного и лицевого нервов). В генетике известны и другие заболевания, обусловленные поражением гена ANKH, в частности – наследственная псевдоподагра или семейный хондрокальциноз.
Намного более редкая аутосомно-рецессивная форма краниометафизарной дисплазии обусловлена дефектом гена GJA1, локализованного на 7-й хромосоме. Продуктом его экспрессии является белок под названием коннексин 43, принимающий активное участие в формировании межклеточных (межщелевых) контактов во многих тканях, что позволяет клеткам обмениваться низкомолекулярными соединениями. Патогенез развития краниометафизарной дисплазии при миссенс-мутации c716G>A неизвестен, основная проблема сводится к выявлению причин изолированного поражения костей черепа и метафизов костей при относительном отсутствии патологий других органов. Учитывая, что наибольшее количество коннексина 43 у человека находится в сердечной ткани, вопрос отсутствия патологий сердца при данной мутации на сегодняшний день представляет собой загадку для большинства врачей-генетиков. Так же, как и в случае аутосомно-доминантной формы краниометафизарной дисплазии, при этом типе заболевания наблюдаются многочисленные вторичные нарушения, обусловленные воздействием измененных костей и отверстий на нервные структуры.
Симптомы краниометафизарной дисплазии
Аутосомно-доминантный тип краниометафизарной дисплазии характеризуется более легким течением, нередко при рождении ребенка никаких симптомов заболевания не выявляется. Лишь к первому году жизни возникает гипертелоризм с расширением переносицы вследствие гиперостоза носовых костей. В дальнейшем краниометафизарная дисплазия приводит к сужению носовых ходов и нарушению их проходимости, поэтому у больных нередко приоткрыт рот из-за нарушенного носового дыхания. К 6-7 годам может начать определяться увеличение метафизов длинных трубчатых костей, что внешне проявляется увеличением размеров коленных и локтевых суставов. Примерно у половины больных краниометафизарной дисплазией развиваются нарушения слуха различной выраженности вплоть до полной глухоты – чаще всего это обусловлено компрессией слухового нерва в костном канале. Из-за сдавления нервов возникают и другие неврологические нарушения, возможны расстройства чувствительности на лице, невралгия тройничного нерва и головные боли.
Краниометафизарная дисплазия аутосомно-рецессивного типа характеризуется намного более выраженными пороками развития костей черепа и конечностей, признаки патологии часто выявляются сразу после рождения ребенка. У больных присутствует выраженный гипертелоризм, черты лица зачастую крайне несимметричны, деформации переносицы и носа могут приобретать признаки уродства. В ряде случаев наблюдаются макроцефалия, нижнечелюстной прогнатизм, другие нарушения прикуса и расположения зубов. По мере роста больного аномалии костей черепа могут усугубляться. Метафизы костей конечностей резко расширены, что нередко обуславливает вторичные деформации (например, Х-образное искривление ног). Как и в предыдущем варианте, при этой форме краниометафизарной дисплазии часто возникают разнообразные неврологические нарушения, вызванные сдавлением и травматизацией черепно-мозговых нервов. Они могут проявляться глухотой, нарушениями зрения, расстройствами кожной чувствительности на лице, парезом мимической мускулатуры и головными болями. Существуют отдельные описания больных, одновременно страдающих краниометафизарной дисплазией и умственной отсталостью, однако достоверных данных о взаимосвязи этих двух состояний на сегодняшний день нет.
Диагностика и лечение краниометафизарной дисплазии
Диагностика краниометафизарной дисплазии основывается на данных осмотра пациента, изучении его наследственного анамнеза, результатах рентгенологических исследований и молекулярно-генетических анализов. При осмотре определяются различные по выраженности аномалии развития костей черепа, что отражается на чертах лица больного. При этом аутосомно-доминантная форма, особенно у маленьких детей, довольно слабо проявляет себя в отношении этого симптома. При обоих типах краниометафизарной дисплазии практически всегда присутствуют гипертелоризм, расширение переносицы за счет разрастания носовых костей по направлению к скулам, сужение или непроходимость носовых ходов. В рамках физикального осмотра и дополнительных исследований могут быть также выявлены неврологические нарушения: снижение или отсутствие слуха, снижение зрения, симптомы со стороны тройничного и лицевого нервов.
Намного больше информации при краниометафизарной дисплазии дают рентгенологические методы исследования скелета. На рентгенограммах черепа при аутосомно-доминантной форме заболевания определяются уплотнение костной ткани в области затылочной кости, склероз основания черепа, пониженная пневматизация синусов и ячеек височной кости. В ряде случаев может выявляться склероз межкостных швов и расширение метафизов трубчатых костей. При аутосомно-рецессивном типе краниометафизарной дисплазии на рентгенограммах обнаруживаются схожие, но намного более выраженные нарушения, например – полное отсутствие околоносовых пазух, резкое сужение и иногда заполнение костной тканью отверстий черепно-мозговых нервов. Кроме того, наблюдается склероз не только основания, но и свода черепа, в ряде случаев определяются значительные деформации костей лицевого отдела. Немного сильнее, чем при доминантном типе, выражены расширение и склероз метафизов трубчатых костей.
Изучение наследственного анамнеза и генетическая диагностика также активно используются для определения краниометафизарной дисплазии. При этом можно сначала установить тип наследования заболевания, что позволяет скорректировать молекулярно-генетический анализ для поиска мутаций в конкретном гене. Для этого применяют метод прямого секвенирования генов ANKH и GJA1.
Специфического лечения краниометафизарной дисплазии не существует, назначают симптоматическую терапию с привлечением разнообразных хирургических техник. Последние, в частности, могут улучшить проходимость носовых ходов для облегчения дыхания. Уменьшить выраженность неврологической симптоматики можно путем расширения отверстий соответствующих черепно-мозговых нервов. Кроме того, пластические хирурги способны минимизировать выраженность эстетических дефектов при краниометафизарной дисплазии. Для улучшения слуха используют слуховые аппараты.
Прогноз и профилактика краниометафизарной дисплазии
Во многих случаях прогноз краниометафизарной дисплазии аутосомно-доминантного типа относительно выживаемости больного благоприятный – ни аномалии черепа, ни вторичные неврологические нарушения не приводят к тяжелым последствиям. Возникающие в детстве патологии слуха или зрения могут медленно прогрессировать до 20-30 лет, после этого ухудшений обычно не происходит. Аутосомно-рецессивный тип краниометафизарной дисплазии характеризуется более неблагоприятным прогнозом, поскольку костные нарушения черепа при этом состоянии имеют тенденцию к усугублению и могут с каждым годом все сильнее травмировать нервы. В некоторых случаях это приводит к параличам и расстройствам со стороны вегетативной нервной системы. Профилактика краниометафизарной дисплазии не разработана, для уменьшения риска развития осложнений при наличии такого состояния необходимо проводить регулярное обследование у невролога.
Гнатодиафизарная дисплазия
Гнатодиафизарная дисплазия – наследственное заболевание скелета, характеризующееся преимущественным поражением нижней челюсти в сочетании с хрупкостью других костей. Симптомами патологии являются частые переломы в детском возрасте с развитием оссифицирующего периодонтита и частых гнойных остеомиелитов челюстей на втором-третьем десятилетии жизни. Диагностика гнатодиафизарной дисплазии производится путем анализа клинической картины и анамнеза жизни пациента, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует.
Гнатодиафизарная дисплазия – генетическая патология, которая приводит к хрупкости костей скелета и нарушению процессов заживления и оссификации нижней челюсти. Впервые это заболевание было описано в 1969 году японским исследователем Акасакой, который выявил у 21-го представителя одного семейства повышенную частоту гнойных остеомиелитов верхней и нижней челюсти. При дальнейшем изучении обнаружилось, что каждый из больных в детском возрасте нередко страдал от переломов костей. Во взрослом возрасте переломы возникали реже, но наблюдались патологии нижней челюсти, иногда с обезображиванием лица. Дальнейшие исследования выявили подобные нарушения и у представителей негроидной расы. Удалось определить, что гнатодиафизарная дисплазия наследуется по аутосомно-доминантному типу, но встречается крайне редко, поэтому ее распространенность до сих пор не определена. Лишь в 2004 году японскими врачами-генетиками Масуко и Масаро Като была выявлена локализация гена, мутации которого приводят к этому заболеванию.
Причины гнатодиафизарной дисплазии
Непосредственной причиной гнатодиафизарной дисплазии являются мутации в гене ANO5, расположенном на 11-й хромосоме. Он кодирует последовательность белка 16Е, который представляет собой трансмембранный протеин из группы кальциевых каналов, способный регулировать практически весь обмен кальция в клетке. Помимо гнатодиафизарной дисплазии, иные мутации в гене ANO5 способны вызывать такие заболевания, как мышечная дистрофия Миоши и поясно-конечностная мышечная дистрофия. Таким образом, эти патологии являются аллельными по отношению к гнатодиафизарной дистрофии. Причины, почему основной объем поражения при данном заболевании приходится на челюстные кости, в настоящий момент неизвестны. Передаются мутации гена ANO5, приводящие к развитию этого заболевания, по аутосомно-доминантному типу.
По поводу патогенеза гнатодиафизарной дисплазии в настоящее время также происходят дискуссии в научном мире. Предполагается, что миссенс-мутация в 11-м экзоне гена ANO5 приводит к изменению трансмембранного белка, что, в свою очередь, затрудняет процессы минерализации за счет нарушения транспорта кальция. В результате этого нарушается структура всех костей (уменьшение толщины коркового слоя, склероз диафизов длинных трубчатых костей, снижение общей плотности костной ткани). Кроме того, во взрослом возрасте, происходит нарушение метаболизма костной ткани в верхней и нижней челюсти. Поэтому часто возникают гнойные остеомиелиты, на месте которых после выздоровления не происходит восстановление нормальной кости, а образуется плотная волокнистая ткань с различной степенью насыщенности цементирующей массой. Аналогичные изменения происходят не только после остеомиелита, но и вследствие переломов челюстных костей.
Симптомы гнатодиафизарной дисплазии
При рождении и в детском возрасте больные гнатодиафизарной дисплазией ничем не отличаются от здоровых сверстников – рост, набор массы, психофизическое развитие остается в пределах нормы. Примерно с возраста 12-13 лет плотность костной ткани начинает падать, но внешне это никак не проявляется. Но типичные для ребенка травматичные ситуации (падения, подвижные игры и другие) нередко оканчиваются переломами. Описаны случаи, когда страдающий гнатодиафизарной дисплазией подросток до 20 лет перенес несколько десятков переломов различных костей. Однако обычно их бывает намного меньше, поэтому родители редко обращают внимание на несколько повышенный травматизм их ребенка, считая это случайным стечением обстоятельств. Ортопед или рентгенолог на снимках переломов может обратить внимание на несколько пониженную плотность костей, но в крайне редких случаях соотносит их с какой-либо наследственной патологией. Срастание переломов происходит в обычные сроки и без различных осложнений.
По достижении 20-летнего возраста у больных гнатодиафизарной дисплазией часто возникают гнойные остеомиелиты верхней и нижней челюсти с их типичной симптоматикой – лихорадкой, симптомами общей интоксикации, сильными болями. На этом этапе возможно развитие таких осложнений, как метастатический перенос гнойного воспаления, токсический шок, сепсис. При благополучном окончании гнойного процесса заживление костной ткани происходит с аномалией – образуется грубая волокнистая ткань с гиперцементозом, соответствующая оссифицирующему периодонтиту. При частых случаях остеомиелита или переломах челюстных костей в результате такой неправильной регенерации может развиваться обезображивание лица. Во взрослом возрасте при гнатодиафизарной дисплазии хрупкость других элементов скелета уменьшается, поэтому частота переломов падает.
Диагностика гнатодиафизарной дисплазии
Диагностика гнатодиафизарной дисплазии производится на основании данных рентгенологического исследования челюстных костей и других элементов скелета, анамнеза жизни пациента, молекулярно-генетических анализов. На рентгене нижней и верхней челюсти определяются очаги, иногда множественные, соответствующие перенесенным остеомиелитам. Они немного прозрачней окружающей костной ткани (по причине наличия там соединительной ткани), но в них имеются участки уплотнения, которые являются гиперцементозом. Иногда, в запущенных случаях гнатодиафизарной дисплазии, отмечаются деформации верхней или нижней челюсти. На рентгенограммах других костей, особенно сделанных в подростковом и молодом возрасте, обнаруживаются снижение толщины кортикального слоя и уменьшение общей плотности кости. Косвенным признаком гнатодиафизарной дисплазии является и наличие следов от многочисленных переломов.
Большое количество травм с переломами костей в детском и подростковом возрасте отмечается и в анамнезе жизни пациента или при его расспросе. Генетическая диагностика сводится к секвенированию последовательности гена ANO5 или только его 11-го экзона. Это позволяет обнаружить миссенс-мутацию, которая и является причиной гнатодиафизарной дисплазии. Кроме того, современная генетика позволяет производить пренатальную диагностику этого заболевания, материал для исследования может быть взят путем амниоцентеза. Дифференциальную диагностику гнатодиафизарной дисплазии следует производить с наследственными и приобретенными формами остеопороза и нарушениями развития костной ткани.
Лечение и прогноз гнатодиафизарной дисплазии
Прогноз гнатодиафизарной дисплазии относительно выздоровления крайне неблагоприятный. Что касается угрозы жизни пациента, то она может быть обусловлена тяжелыми переломами в детском и подростковом возрасте, а также осложнениями гнойного остеомиелита. В большинстве случаев больные доживают до преклонного возраста, особенно при своевременном обращении к врачу при различных патологических процессах. Иногда может возникнуть обезображивание лица из-за деформаций челюстных костей, которое может устраняться методами пластической хирургии.
Читайте также:
- Лечение эндогенной интоксикации. Интенсивная терапия эндогеннной интоксикации.
- Симптомы декомпрессионной болезни и его лечение
- Опухолеподобные поражения щитовидной железы. Эктопическая тиреоидная ткань.
- Рентгенограмма, КТ, МРТ при нейрофиброматозе
- Взаимодействия антителообразующих клеток. Влияние клеток на синтез антител