Лечение при постполицитемическом миелофиброзе с миелоидной метаплазией (МММ) - эффективность
Добавил пользователь Skiper Обновлено: 28.01.2026
Лечение при постполицитемическом миелофиброзе с миелоидной метаплазией (МММ) - эффективность
Лечение в постэритремической стадии истинной полицитемии (ИП) определяется характером гематологического исхода. В целом это миелофиброз с миелоидной метаплазией (МММ), но с различной величиной селезенки, различием гематологических данных и гистоморфологии костного мозга — от гиперклеточного до диффузно-фиброзного.
Принципы лечения в стадии постэритремического миелофиброза с миелоидной метаплазией (МММ) аналогичны таковым при ХИМФ, с тем отличием, что при первом могут сохраняться повышенные показатели красной крови и, следовательно, необходимость в эксфузиях крови. Хотя опасность лейкозогенного действия цитостатических препаратов в этой стадии резко возрастает, их приходится назначать для контроля над прогрессирующим лейкоцитозом, нарастающим числом промежуточных форм белого ряда и спленомегалией. Большого опыта терапии ИФН-а этой стадии истинной полицитемии (ИП) еще нет, и вряд ли он будет очень успешным.
Следует отметить, что клинические и гематологические проявления миелофиброза с миелоидной метаплазией (МММ) неоднозначны. У целого ряда больных темпы роста селезенки весьма умеренны, анемии нет и предметом коррекции оказывается один тромбоцитоз. Возможно, что для таких больных средством выбора будет анагрелид, но нами применялась Hydrea в течение длительного срока с хорошей переносимостью и эффектом.
По опыту отечественной гематологии, лейкозогенное действие Hydrea несравненно меньшее, чем у алкилирующих агентов, назначения которых в этой стадии действительно лучше избегать.
Для больных, завершивших эритремическую стадию периодом стабилизации, активная терапия может и вовсе не потребоваться.
Терапевтическая тактика в случаях прогрессирующего увеличения селезенки при отсутствии лейкоцитоза и ромбоцитоза проблематична. Попытки применения Hydrea в небольших дозах приносят малый эффект. Терапевтические возможности ИФН-а в подобной ситуации пока не определены. Нужно помнить, что прогрессирование спленомегалии может быть обусловлено не только нарастанием экстрамедуллярного кроветворения, но и внутрипеченочной портальной гипертонией (ПГ), обычно без больших анатомических предпосылок для этого.
Это так называемый функциональный блок в печени, что подтверждают результаты секционного исследования печени у умерших от пищеводных кровотечений. Больные с данным осложнением подлежат спленэктомии (СЭ), которая, однако, не гарантирует дальнейшего гематологического благополучия.
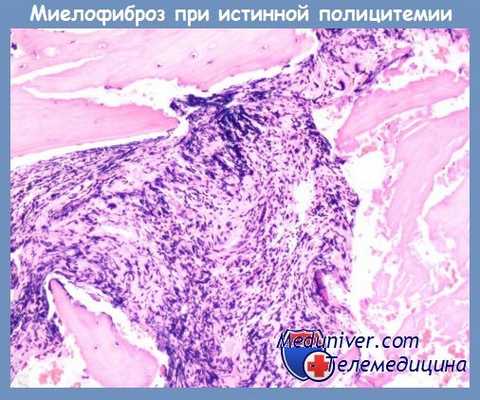
Лечение анемического синдрома в этой стадии осуществляется с учетом патогенеза анемии. Оно тождественно его лечению при ХИМФ, включая назначение спленэктомии (СЭ) при гемолитическом характере анемии.
Лечение анемии, вызванной нарушенным образованием эритроцитов в результате замещения кроветворного костного мозга диффузным миелофибро-зом, или неэффективного эритропоэза, или смены ведущей эритроидной линии пролиферации на гра-нулоцитарную, является наиболее трудной задачей. Это в полной мере относится и к рефрактерной си-деробластной анемии, предстадии острого лейкоза. Любая из указанных выше предпосылок к развитию анемии, в сущности, относится к числу терминальных состояний и плохо поддается коррекции, в том числе андрогенами и цитостатиками, глюкокортико-стероидами и ИФН-а.
Попытки лечения макроцитарных анемий фолиевой кислотой нами неоднократно предпринимались, но почти всегда безуспешно. Назначение препаратов железа при оставшемся от плеторической стадии его дефиците оправдано, если дефицит железа подтвержден исследованием уровня феррити-на. Показатели транспортного фонда железа часто дают ошибочные результаты.
Лечение тромбоцитопении также определяется причиной ее развития. Как и при ХИМФ, она является показанием к спленэктомии, если обусловлена гиперспленизмом. К тромбоцитопении может привести предшествующая цитостатическая терапия, и особенно терапия миелосаном. В подобных случаях спленэктомия не показана. Тромбоцитопения при малом числе мегакариоцитов в костном мозге и при преобладании мелких и атипичных по структуре форм чаще всего является проявлением миелодисплазии. В этих случаях спленэктомия неэффективна и не должна назначаться.
Опыт спленэктомии центра РАМН включает 12 больных. Повышенной кровоточивостью сопровождались практически все спленэктомии. Осложнения тромбофлебитами наблюдались у 2/3 пациентов почти у всех имелся тромбоцитоз в послеоперационном периоде. Двое больных погибли от осложнений, связанных с операцией. Причины смерти 2 больных — миелодисплазия, у 3 — острый лейкоз.
Продолжительность жизни у перенесших спленэктомию больных составила 1,5 года — 8 лет.

При анализе отдаленных результатов спленэктомии у нас не сложилось впечатления, что операция явилась причиной развития острого лейкоза, скорее, она не предотвратила его развития. Целесообразность спленэктомии в случаях миелодисплазии крайне сомнительна; когда же больные оперируются по поводу компрессионного синдрома при гематологической стабильности, портальной гипертонии, гемолитической анемии, достигаются хорошие результаты.
В постэритремической стадии возрастает необходимость в симптоматическом лечении. Объектами этой терапии становятся истощение больного, при котором показаны анаболические гормоны, гиперкатаболический синдром, когда полезными оказываются кортикостероиды (преднизолон в суточной дозе 10—15 мг), уратовый диатез, при котором необходима терапия аллопуринолом, и сосудистые тромбофилические и геморрагические осложнения. Для симптоматической терапии массивной спленомегалии (в случаях, когда спленэктомия противопоказана) применяется гамма-терапия.
Прежде она назначалась часто, но получаемые результаты были кратковременными (в среднем 7—8 месяцев), цитопенические осложнения — нередкими, темпы роста селезенки после ее временного сокращения — возрастающими. Это объясняет общее крайне сдержанное отношение к данному методу лечения.
Лечение острого лейкоза у больных истинной полицитемией (ИП) проводится по общим правилам и иногда успешно.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Интерферон (ИФН-а) в лечении истинной полицитемии - эффективность
Новым направлением в лечении истинной полицитемии (ИП) является терапия рекомбинантным ИФН-а. В основе лечебного действия данного препарата лежит опосредованное цитокинами подавление патологической миелопролиферации, приводящее к снижению числа тромбоцитов, лейкоцитов и эритроцитов. Он является также ингибитором продуцируемого тромбоцитами ростового фактора (PDGF-b), поэтому можно ожидать его действия, сдерживающего развитие миелофиброза. Доказана способность препарата подавлять рост эритроидных предшественников in vivo и in vitro, а также вызывать морфологические изменения в мегакариоцитах.
За критерий полной ремиссии у леченных ИФН-а принято отсутствие необходимости в кровопусканиях для поддержания нормального Ht, частичной — уменьшение потребности в кровопусканиях на 50 % и более. Дополнительно оценивается влияние на число тромбоцитов, лейкоцитов, величину селезенки.
Другой анализ обобщенных результатов более поздних исследований и с большей продолжительностью лечения показал, что в 60 % случаев достигнута полная ремиссия, в 70 % — редукция увеличенной селезенки, а в 75 % — контроль над кожным зудом.
Немецкие авторы сообщили о суммарных результатах лечения 279 больных из 16 нерандомизированных исследований и 3 собственных больных с большими сроками лечения (до 126 мес в отдельных случаях). Начальная доза ИФН-а варьировала от 3 до 35 ME в неделю. Полная ремиссия получена у 50 % больных и в последующем поддерживалась меньшими дозами ИФН-а. В 21 % случаев лечение было прервано из-за осложнений.
Детальный анализ влияния ИФН-а на отдельные параметры заболевания представлен в работе Н. Gilbert и соавт.. В период индукции ремиссии препарат назначался по 30 ME в неделю, а в период закрепления результатов — в половину меньшей дозе. Достигнута нормализация числа тромбоцитов у всех больных, снижение числа лейкоцитов на 20 % от исходного уровня в 48 %, уменьшение размеров селезенки в 73 %. Влияние на показатели красной крови в работе не обсуждается. Результаты лечения зависели от стадии заболевания. Так, у больных с ММС селезенка сократилась лишь в 19 %, а в стадии пост-полицитемической ММС — в 0,1 % случаев.
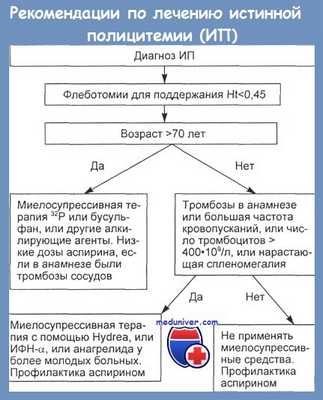
Принято считать, что прекращение лечения приводит к быстрому рецидиву заболевания, но авторами показано, что у 72 % больных по окончании лечения достигнутый эффект сохранялся в среднем в течение 39 нед, а у отдельных больных — до 2 лет. Повторные курсы лечения у больных, ответивших на первый курс, были столь же эффективными. Если эти данные найдут дальнейшее подтверждение, стратегия лечения ИФН-а истинной полицитемии может измениться.
Большинство авторов обычно начинают лечение с 9 ME в неделю и через 1—3 мес при необходимости увеличивают дозу вдвое, продолжая наращивать дозы и далее, но не более чем до 30 ME в неделю.
Показательны результаты лечения истинной полицитемии низкими дозами ИФН-а в Mayo-Clinic, США. Автор обобщил опыт лечения 55 больных истинной полицитемией за последние 15 лет. Средний возраст больных составил 49 лет. Продолжительность заболевания до начала лечения — от 33 до 205 мес, максимальная — 356 мес. Схема лечения включала первоначальное назначение кровопусканий до нормализации Ht (40—45 %). Затем назначался ИФН-а по 1 ME 2 раза в неделю подкожно. Эскалация дозы производилась постепенно до 3 ME 2 раза в неделю. При ухудшении переносимости вводимая доза уменьшалась до хорошо переносимой дозы.
Через 1 год полная ремиссия, которую автор определил как отсутствие потребности в кровопусканиях и число тромбоцитов менее 600•109/л, была достигнута у всех больных. Четверо больных нуждались в 1—2 кровопусканиях в год. До лечения ИФН-а потребность в кровопусканиях составляла до 70 в год. Поддерживающая доза в этом исследовании — 2—3 ME 2 раза в неделю. Средняя продолжительность лечения составила от 12 до 101 мес, самая большая — 163 мес (около 14 лет).
Лечение было прекращено у 15 больных в возрасте старше 55 лет по разным причинам, но только у 10 — из-за поздней токсичности (тяжелый дерматит, комплекс осложнений, периферический неврит и др.). Повторные трепанобиопсии костного мозга выявляли его гиперклеточность, и только у 3 больных через 18, 25 и 30 лет от начала заболевания обнаружен «мягкий» миелофиброз с небольшим увеличением селезенки. Случаев острого лейкоза не было.
В другом исследовании также отмечается сохранение гиперплазии костного мозга у леченных ИФН-а, тогда как у принимавших Hydrea и бусульфан отмечено ее существенное уменьшение. Эти данные свидетельствуют о принципиально ином механизме действия ИФН-а по сравнению с цитостатическими препаратами.
Дальнейшие наблюдения покажут, способна ли эта терапия предупреждать эволюцию истинной полицитемии в миелофиброз с миелоидной метаплазией (МММ) и вызывать его реверсию. При гистоморфологическом исследовании костного мозга у 3 наших больных накануне лечения миелофиброз был обнаружен у одного, а через 1 год лечения — у всех 3 больных, но все они имели большую продолжительность заболевания и миелоидной метаплазии селезенки (ММС). С учетом приведенных выше данных имеются основания ожидать урежения и отдаления сроков развития миелофиброза с миелоидной метаплазией (МММ).
В одном исследовании у 2 больных истинной полицитемией с исходной цитогенетической патологией она исчезла под влиянием лечения. Эти данные подтверждают способность ИФН-а вызывать эрадикацию неопластического клона.
Особенно интересна способность ИФН-а подавлять кожный зуд, связанный с приемом водных процедур, поскольку другие виды циторедуктивной терапии не дают подобного эффекта. Длительность терапии, приведшей к исчезновению кожного зуда, составила 11 — 12 мес. При исследовании экскреции гистамина у больных, леченных ИФН-а, авторы не нашли ее уменьшения и на этом основании высказали сомнения в достоверности предполагаемой роли гистаминемии в патогенезе кожного зуда.
Лечение ИФН-а сопровождается ранними и более опасными поздними осложнениями. К последним относят потерю массы тела (до 20 кг за 1 год при дозе в 30 ME в неделю), психические нарушения вплоть до депрессии, нарушения функций печени и почек, иногда инфекционные и другие осложнения. Появление осложнений вынуждает прекратить лечение у 10—20 % больных.
Миелодиспластический синдром
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Общие сведения
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Миелофиброз ( Агногенная миелоидная метаплазия , Сублейкемический миелоз )
Миелофиброз – это хроническое гематологическое заболевание, характеризующееся опухолевой пролиферацией гемопоэтических стволовых клеток и фиброзом костного мозга. Основные клинические проявления включают симптомы опухолевой интоксикации и анемического синдрома (прогрессирующую слабость, бледность кожи и слизистых оболочек, потерю веса), а также увеличение селезенки (спленомегалию). Диагноз устанавливается на основании молекулярно-генетических исследований, изучения гистологической картины костного мозга. Лечение проводится с помощью химиотерапевтических препаратов. Хирургические методы лечения подразумевают трансплантацию костного мозга и удаление селезенки.
МКБ-10
Миелофиброз (агногенная миелоидная метаплазия, сублейкемический миелоз) – злокачественное заболевание, при котором происходит постепенное замещение костного мозга опухолевыми стволовыми клетками и разрастающейся соединительной тканью. Впервые эту патологию описал немецкий врач Г. Хойк в 1879 году. А в 1951 году американским гематологом Уильямом Дамешеком миелофиброз был выделен в самостоятельную нозологическую единицу. При неблагоприятном течении миелофиброз способен трансформироваться в более тяжелую болезнь ‒ острый лейкоз. Распространенность миелофиброза составляет от 0,3 до 0,7 случаев на 100 тыс. населения. Пик заболеваемости приходится на возраст от 50 до 70 лет, но встречаются и молодые пациенты. Чаще страдают мужчины.
Причины миелофиброза
Существует первичный и вторичный сублейкемический миелоз. Точная причина первичного миелофиброза до сих пор не установлена. Наибольшей популярностью среди специалистов в области гематологии пользуется теория влияния генетической мутации. У большинства пациентов выявляются мутации гена тирозинкиназы (JAK2V617F), кальретикулина (CALR), тромбопоэтина (MPL), регулирующих экспрессию белков JAK-STAT сигнального пути. Гены локализуются в локусе хромосомы del3p24.
В качестве этиологического фактора изучается действие большой дозы радиоактивного излучения. Также рассматривается роль персистирующих вирусных инфекций (вируса простого герпеса, Эпштейна-Барра, цитомегаловируса), длительного приема оральных контрацептивов, миелосупрессивных лекарственных препаратов, контакта с различными органическими и неорганическими соединениями (бензолом, мышьяком). Вторичный миелофиброз развивается как исход других хронических миелопролиферативных заболеваний – истинной полицитемии, эссенциальной тромбоцитемии, хронического миелолейкоза.
Патогенез
В результате повышенной экспрессии сигнальных белков в одной из стволовых костномозговых клеток запускается активная пролиферация (опухолевая трансформация). Этот процесс сопровождается вторичным воспалением с выделением цитокинов и факторов роста. Факторы роста фибробластов и эндотелия сосудов индуцируют выработку стромальными клетками костного мозга большого количества коллагена и разрастание соединительной ткани (собственно фиброз). Постепенно нормальная ткань костного мозга замещается опухолью и соединительной тканью.
При массивном поражении опухолью костного мозга клетки крови, не достигнув стадии полного созревания, попадают в системный кровоток. Это приводит к образованию очагов экстрамедуллярного (внекостномозгового) кроветворения, главным образом в печени и селезенке. Распад опухоли ведет к высвобождению мочевой кислоты, которая откладывается в тканях суставов и почечных канальцах.
Симптомы миелофиброза
Длительное время пациент чувствует себя удовлетворительно. Через несколько лет от начала заболевания постепенно появляется опухолевая интоксикация в виде общей слабости, повышения температуры до субфебрильных цифр, потливости, усиливающейся по ночам. У больного снижается аппетит, он стремительно теряет в весе. Присоединяется анемический синдром (бледность кожных покровов, головокружение, учащение сердцебиения). Характерны носовые, десневые кровотечения, геморрагические высыпания на коже. Возникают боли в суставах, кожный зуд, боли в костях.
Пациент ощущает тяжесть и боли в левом подреберье вследствие выраженного увеличения селезенки. На фоне спленомегалии развивается синдром гиперспленизма, который заключается в массивном разрушении клеток крови (в основном эритроцитов) в синусоидах селезенки. В этом случае встречаются признаки гемолиза (желтушность кожи, слизистых оболочек, потемнение мочи).
Редкие симптомы связаны с необычной локализацией очагов экстрамедуллярного кроветворения – в легких (кашель, затруднение дыхания, кровохарканье), желудочно-кишечном тракте (боли в животе, кровавая диарея). При расположении очагов в центральной и периферической нервной системе наблюдаются эпилептические судороги, нарушения чувствительности, слабость движений в конечностях, вплоть до полного паралича.
Осложнения
При миелофиброзе часто образуются тромбы, которые приводят к острому нарушению мозгового кровообращения, инфаркту миокарда, тромбоэмболии легочной артерии. Стойкое снижение уровня лейкоцитов нередко сопряжено с различными инфекциями, приобретающими тяжелое течение. Наиболее неблагоприятным осложнением считается трансформация миелофиброза в миелолейкоз (бластный криз), трудно поддающийся терапии. К нетипичным осложнениям следует отнести патологические переломы из-за деструкции трубчатых костей и портальную гипертензию, причиной которой служит длительная обструкция микротромбами внутрипеченочных вен.
Диагностика
Курацией пациентов с миелофиброзом занимаются врачи-гематологи. При общем осмотре обращает на себя внимание изменение цвета кожных покровов, слизистых (бледность или желтушность), спленомегалия при пальпации и перкуссии селезенки, иногда достигающей гигантских размеров (до лобкового симфиза). Дополнительные методы диагностики включают:
- Общие лабораторные исследования. В начале заболевания в общем анализе крови выявляется увеличение эритроцитов, тромбоцитов, лейкоцитов, со временем сменяющееся на низкие показатели. Часто в периферической крови присутствуют незрелые формы эритроцитов, лейкоцитов (миелоциты, промиелоциты). В биохимическом анализе крови наблюдаются повышенные концентрации лактатдегидрогеназы (ЛДГ), ионизированного кальция. Отмечаются изменения коагулограммы – ускорение свертывания крови, уменьшение активированного частичного тромбопластинового времени, торможение процессов фибринолиза. В анализе мочи обнаруживаются уробилин, гемоглобин, ураты (соли мочевой кислоты).
- Исследование костного мозга. Образец костного мозга получают с помощью трепанобиопсии. Гистологическая картина зависит от фазы заболевания. Для ранней (префибротической фазы) характерны гиперплазия всех ростков кроветворения (гранулоцитарного, мегакариоцитарного, эритроидного) с незрелостью клеток. В позднюю (фибротическую) фазу определяется большое количество коллагеновых и ретикулярных волокон (фиброз), замещающих гемопоэтическую ткань, выраженная клеточная атипия. Высокий уровень бластных клеток (более 20%) свидетельствует о трансформации миелофиброза в острый лейкоз.
- Молекулярно-генетические тесты. Диагностика мутации генов JAK2V617F, CALR, MPL осуществляется методом FISH. Для идентификации аллельной нагрузки мутации проводится полимеразная цепная реакция real-time. Также выполняется HLA-типирование для решения вопроса о возможности трансплантации костного мозга.
- Цитогенетические и цитохимические анализы. При цитогенетическом исследовании (кариотипировании) клеток костного мозга находят аномалии 1, 3, 6 хромосом (транслокация, трисомия, комплексные нарушения). При анализе химического состава (цитохимии) нейтрофилов активность щелочной фосфатазы оказывается в 3 раза выше нормы.
Для достоверной постановки диагноза гематологическим сообществом разработаны специальные критерии. Большие критерии включают повышенную клеточность костного мозга с ретикулярным и коллагеновым фиброзом, наличие мутаций генов JAK2V617F, MPL, CALR. К малым критериям относятся анемия, спленомегалия, лейкоэритробластоз (присутствие в крови незрелых форм лейкоцитов, эритроцитов), а также повышение лактатдегидрогеназы. Диагноз считается подтвержденным, если имеются 2 больших критерия или 1 большой и 3 малых критерия.
Миелофиброз следует дифференцировать в первую очередь с гематологическими заболеваниями, такими как аутоиммунные гемолитические анемии, гемобластозы (лейкозы, лимфомы). Сочетание спленомегалии с симптомами интоксикации (слабостью, субфебрилитетом, ночной потливостью) требует исключения туберкулеза, подострого инфекционного эндокардита.
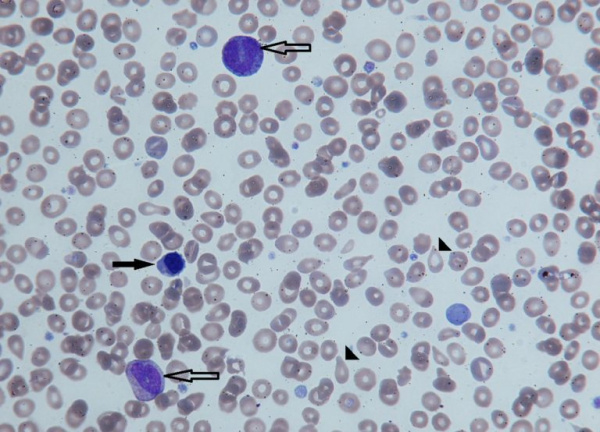
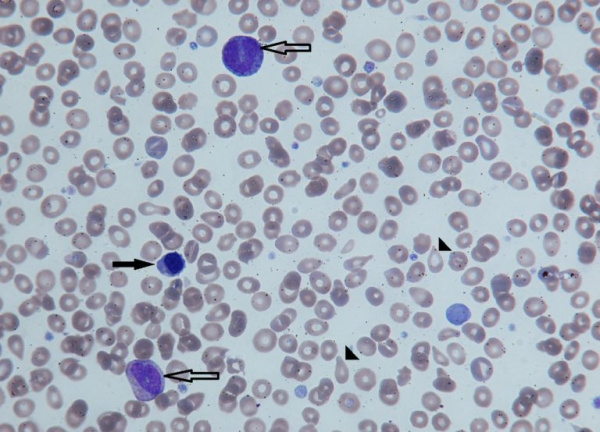
Незрелые формы эритроцитов (черная стрелка) и гранулоцитов (контурная стрелка) в периферической крови
Лечение миелофиброза
После постановки диагноза пациент должен быть госпитализирован в гематологический стационар. Для принятия решения о выборе тактики лечения необходимо определить степень риска, а именно - вероятность бластной трансформации и ориентировочную продолжительность жизни. С этой целью была создана «Международная шкала оценки риска и прогноза» (DIPSS). Она учитывает возраст пациента, количество форменных элементов крови, выраженность симптомов опухолевой интоксикации. Каждый признак соответствует одному баллу. Различают низкий, первый и второй промежуточный, высокий риски, при которых проводится дифференцированная терапия:
Прогноз и профилактика
Миелофиброз – это тяжелое заболевание с неблагоприятным прогнозом. С момента постановки диагноза средняя продолжительность жизни составляет около 5 лет. При манифестации в более молодом возрасте миелофиброз имеет менее агрессивное течение, что сопряжено с лучшим ответом на терапию и большей выживаемостью больных. Эффективных методов профилактики не разработано ввиду неизвестности этиологического фактора. Предупреждение развития вторичного миелофиброза заключается в своевременной диагностике и лечении патологий, на фоне которых он возникает - истинной полицитемии и эссенциальной тромбоцитемии.
2. Патофизиологические основы лечения сублейкемического миелоза. Патофизиология крови. Экстремальные состояния/ Под ред. А.И. Воробьева и Н.А. Горбуновой - 2004.
3. Критерии диагностики и современные методы лечения первичного миелофиброза/ Абдулкадыров К. М., Шуваев В. А., Мартынкевич И. С.// Вестник гематологии. - 2013 - №9(3).
4. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитопения, первичный миелофиброз. - 2014.
Острый миелоидный лейкоз
Острый миелоидный лейкоз – злокачественное заболевание системы крови, сопровождающееся неконтролируемым размножением измененных лейкоцитов, снижением количества эритроцитов, тромбоцитов и нормальных лейкоцитов. Проявляется повышенной склонностью к развитию инфекций, лихорадкой, быстрой утомляемостью, потерей веса, анемией, кровоточивостью, образованием петехий и гематом, болями в костях и суставах. Иногда выявляются изменения кожи и припухлость десен. Диагноз устанавливается на основании клинических симптомов и данных лабораторных исследований. Лечение – химиотерапия, трансплантация костного мозга.
Острый миелоидный лейкоз (ОМЛ) – злокачественное поражение миелоидного ростка крови. Неконтролируемая пролиферация лейкозных клеток в костном мозге влечет за собой подавление остальных ростков крови. В результате количество нормальных клеток в периферической крови уменьшается, возникают анемия и тромбоцитопения. Острый миелоидный лейкоз является самым распространенным острым лейкозом у взрослых. Вероятность развития болезни резко увеличивается после 50 лет. Средний возраст пациентов составляет 63 года. Мужчины и женщины молодого и среднего возраста страдают одинаково часто. В старшей возрастной группе наблюдается преобладание лиц мужского пола. Прогноз зависит от вида острого миелоидного лейкоза, пятилетняя выживаемость колеблется от 15 до 70%. Лечение осуществляют специалисты в области онкологии и гематологии.
Причины острого миелоидного лейкоза
Непосредственной причиной развития ОМЛ являются различные хромосомные нарушения. В числе факторов риска, способствующих развитию таких нарушений, указывают неблагоприятную наследственность, ионизирующее излучение, контакт с некоторыми токсическими веществами, прием ряда лекарственных препаратов, курение и болезни крови. Вероятность возникновения острого миелоидного лейкоза увеличивается при синдроме Блума (низкий рост, высокий голос, характерные черты лица и разнообразные кожные проявления, в том числе гипо- или гиперпигментация, кожная сыпь, ихтиоз, гипертрихоз) и анемии Фанкони (низкий рост, дефекты пигментации, неврологические расстройства, аномалии скелета, сердца, почек и половых органов).
Острый миелоидный лейкоз достаточно часто развивается у больных с синдромом Дауна. Прослеживается также наследственная предрасположенность при отсутствии генетических заболеваний. При ОМЛ у близких родственников вероятность возникновения болезни повышается в 5 раз по сравнению со средними показателями по популяции. Самый высокий уровень корреляции выявляется у однояйцевых близнецов. Если острый миелоидный лейкоз диагностируется у одного близнеца, риск у второго составляет 25%. Одним из важнейших факторов, провоцирующих ОМЛ, являются заболевания крови. Хронический миелоидный лейкоз в 80% случаев трансформируется в острую форму болезни. Кроме того, ОМЛ нередко становится исходом миелодиспластического синдрома.
Ионизирующее излучение вызывает острые миелоидные лейкозы при превышении дозы 1 Гр. Заболеваемость увеличивается пропорционально дозе облучения. На практике имеет значение пребывание в зонах атомных взрывов и аварий на атомных электростанциях, работа с источниками излучения без соответствующих защитных средств и радиотерапия, применяемая при лечении некоторых онкологических заболеваний. Причиной развития острого миелоидного лейкоза при контакте с токсическими веществами является аплазия костного мозга в результате мутаций и поражения стволовых клеток. Доказано негативное влияние толуола и бензола. Обычно ОМЛ и другие острые лейкозы диагностируются спустя 1-5 лет после контакта с мутагеном.
В числе лекарственных средств, способных провоцировать острые миелоидные лейкозы, специалисты называют некоторые препараты для химиотерапии, в том числе ингибиторы ДНК-топоизомеразы II (тенипозид, этопозид, доксорубицин и другие антрациклины) и алкилирующие средства (тиофосфамид, эмбихин, циклофосфамид, хлорамбуцил, кармустин, бусульфан). ОМЛ также может возникать после приема хлорамфеникола, фенилбутазона и препаратов мышьяка. Доля лекарственных острых миелоидных лейкозов составляет 10-20% от общего количества случаев заболевания. Курение не только повышает вероятность развития ОМЛ, но и ухудшает прогноз. Средняя пятилетняя выживаемость и продолжительность полных ремиссий у курильщиков ниже, чем у некурящих.
Классификация острого миелоидного лейкоза
Классификация острого миелоидного лейкоза по версии ВОЗ очень сложна и включает в себя несколько десятков разновидностей заболевания, разделенных на следующие группы:
- ОМЛ с типичными генетическими изменениями.
- ОМЛ с изменениями, обусловленными дисплазией.
- Вторичные острые миелоидные лейкозы, возникшие в результате лечения других заболеваний.
- Болезни с пролиферацией миелоидного ростка при синдроме Дауна.
- Миелоидная саркома.
- Бластная плазмацитоидная дендритноклеточная опухоль.
- Другие виды острого миелоидного лейкоза.
Тактика лечения, прогноз и продолжительность ремиссий при разных видах ОМЛ могут существенно различаться.
Симптомы острого миелоидного лейкоза
Клиническая картина включает в себя токсический, геморрагический, анемический синдромы и синдром инфекционных осложнений. На ранних стадиях проявления острого миелоидного лейкоза неспецифичны. Отмечается повышение температуры без признаков катарального воспаления, слабость, утомляемость, потеря веса и аппетита. При анемии присоединяются головокружения, обморочные состояния и бледность кожных покровов. При тромбоцитопении наблюдаются повышенная кровоточивость и петехиальные кровоизлияния. Возможно образование гематом при незначительных ушибах. При лейкопении возникают инфекционные осложнения: частые нагноения ран и царапин, упорные повторные воспаления носоглотки и т. д.
В отличие от острого лимфобластного лейкоза при остром миелоидном лейкозе отсутствуют выраженные изменения со стороны периферических лимфатических узлов. Лимфоузлы небольшие, подвижные, безболезненные. Иногда выявляется увеличение лимфатических узлов в шейно-надключичной области. Печень и селезенка в пределах нормы или незначительно увеличены. Характерны признаки поражения костно-суставного аппарата. Многие больные острым миелоидным лейкозом предъявляют жалобы на боли различной степени интенсивности в области позвоночника и нижних конечностей. Возможны ограничения движений и изменения походки.
В числе экстрамедуллярных проявлений острого миелоидного лейкоза – гингивит и экзофтальм. В отдельных случаях наблюдаются припухлость десен и увеличение небных миндалин в результате инфильтрации лейкозными клетками. При миелоидной саркоме (составляет около 10% от общего количества случаев острого миелоидного лейкоза) на коже пациентов появляются зеленоватые, реже – розовые, серые, белые или коричневые опухолевидные образования (хлоромы, кожные лейкемиды). Иногда при поражениях кожи обнаруживается паранеопластический синдром (синдром Свита), который проявляется воспалением кожных покровов вокруг лейкемидов.
В развитии острого миелоидного лейкоза выделяют пять периодов: начальный или доклинический, разгара, ремиссии, рецидива и терминальный. В начальном периоде острый миелоидный лейкоз протекает бессимптомно или проявляется неспецифической симптоматикой. В периоде разгара токсический синдром становится более выраженным, выявляются анемический, геморрагический и инфекционный синдромы. В период ремиссии проявления острого миелоидного лейкоза исчезают. Рецидивы протекают аналогично периоду разгара. Терминальный период сопровождается прогрессирующим ухудшением состояния больного и завершается летальным исходом.
Диагностика и лечение острого миелоидного лейкоза
Решающую роль в процессе диагностики играют лабораторные анализы. Используют анализ периферической крови, миелограмму, микроскопические и цитогенетические исследования. Для получения образца тканей выполняют аспирационную биопсию костного мозга (стернальную пункцию). В анализе периферической крови больного острым миелоидным лейкозом обнаруживается снижение количества эритроцитов и тромбоцитов. Количество лейкоцитов может быть как повышенным, так и (реже) пониженным. В мазках могут выявляться бласты. Основанием для постановки диагноза «острый миелоидный лейкоз» становится обнаружение более 20% бластных клеток в крови либо в костном мозге.
Основой лечения острого миелоидного лейкоза является химиотерапия. Выделяют два этапа лечения: индукцию и консолидацию (постремиссионную терапию). На этапе индукции выполняют лечебные мероприятия, направленные на уменьшение количества лейкозных клеток и достижение состояния ремиссии. На этапе консолидации устраняют остаточные явления болезни и предотвращают рецидивы. Лечебную тактику определяют в зависимости от вида острого миелоидного лейкоза, общего состояния больного и некоторых других факторов.
Наиболее популярная схема индукционного лечения – «7+3», предусматривающая непрерывное внутривенное введение цитарабина в течение 7 дней в сочетании с одновременным быстрым периодическим введением антрациклинового антибиотика в течение первых 3 дней. Наряду с этой схемой в процессе лечения острого миелоидного лейкоза могут применяться другие лечебные программы. При наличии тяжелых соматических заболеваний и высоком риске развития инфекционных осложнений в результате подавления миелоидного ростка (обычно – у больных старческого возраста) используют менее интенсивную паллиативную терапию.
Программы индукции позволяет добиться ремиссии у 50-70% пациентов с острым миелоидным лейкозом. Однако без дальнейшей консолидации у большинства больных наступает рецидив, поэтому второй этап лечения рассматривается, как обязательная часть терапии. План консолидационного лечения острого миелоидного лейкоза составляется индивидуально и включает в себя 3-5 курсов химиотерапии. При высоком риске рецидивирования и уже развившихся рецидивах показана трансплантация костного мозга. Другие методы лечения рецидивных ОМЛ пока находятся в стадии клинических испытаний.
Прогноз острого миелоидного лейкоза
Прогноз определятся разновидностью острого миелоидного лейкоза, возрастом больного, наличием или отсутствием миелодиспластического синдрома в анамнезе. Средняя пятилетняя выживаемость при разных формах ОМЛ колеблется от 15 до 70%, вероятность развития рецидивов – от 33 до 78%. У пожилых людей прогноз хуже, чем у молодых, что объясняется наличием сопутствующих соматических заболеваний, являющихся противопоказанием для проведения интенсивной химиотерапии. При миелодиспластическом синдроме прогноз хуже, чем при первичном остром миелоидном лейкозе и ОМЛ, возникшем на фоне фармакотерапии по поводу других онкологических заболеваний.
Читайте также:
- Лечение острой сердечной недостаточности. Трансформация острой сердечной недостаточности.
- Ногти. Строение ногтей. Реактивность и регенерация кожи и ее производных.
- Клиника трихомоноза. Диагностика трихомоноза. Лечение трихомоноза.
- Семейная отогенная интоксикация стрептомицином. Синдром Тернера с глухотой
- Обзор бесплодия