Механизмы развития метгемоглобинемии - патофизиология
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Метгемоглобинемии - история изучения, причины
Метгемоглобинемии составляют разнородную группу наследственных или приобретенных заболеваний, которые характеризуются наличием в крови увеличенного количества метгемоглобина. Метгемоглобин (МетГб) это окисленная форма (Fe3+) гемоглобина, непригодного для выполнения ведущей функции переноса кислорода. С клинической точки зрения метгемоглобинемия проявляется цианозом, признаками недостатка кислорода, а иногда гемолитической анемией с тельцами Гейнца, или без таковых.
К этой группе заболеваний относятся как приобретенная метгемоглобинемия, при которой образование МетГб происходит под воздействием случайно вводимых в организм метгемоглобинизирующих веществ или медикаментов, так и наследственная метгемоглобинемия, в основу которой заложена генетическая аномалия структуры гемоглобина или набора ферментов эритроцита.
Первый случай наследственной метгемоглобинемии, за счет аномалии гемоглобина, описан Horlein и Weber в 1948 г. в одной немецкой семье. К тому времени молекулярные болезни еще не были известны и аномальный гемоглобин не вскрыт. Лишь в 1961 г. Gerald и Efron описали первые случаи гемоглобина М и механизм образования метгемоглобина в связи с структурной аномалией молекулы гемоглобина.
Этот факт сделал возможным не только дифференциацию приобретенной (токсической природы) и наследственной метгемоглобинемии, но и выделение в отдельные единицы метгемоглобинемии ферментативной природы и метгемоглобинемии за счет наличия гемоглобина М.
Причины (этиология) метгемоглобинемии
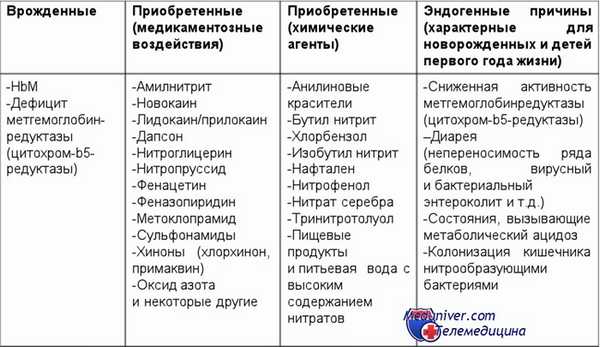
Наиболее часто встречается приобретенная метгемоглобинемия. Причины — многочисленны и разнообразны, они включают совокупность тех положений, когда в организм поступают химические метгемоглобинизирующие вещества в достаточно большом количестве и тем самым преодолевают восстановительную способность эритроцита.
Ниже приведены токсические агенты, наиболее часто встречающиеся на практике. При этом некоторые из них — средства текущей терапевтической практики, в то время как другие — вводятся в организм случайно. Наиболее активные — производные анилина, сульфонамиды, нитриты.
Потребление колодезной воды,зараженной нитритами (преобразуемыми кишечной флорой в нитраты) или случайное заглатывание карандаша или мела, содержащих анилиновые красители — относительно часто встречаются, особенно в педиатрии. Описан случай энтеро-генного цианоза с метгемоглобином и сульфгемоглобином после повышенного поглощения кишечником нитритов и соединений серы, видимо в связи с нарушением функции кишок.
Причины, вызывающие метгемоглобинемию:
I. Наследственные причины метгемоглобинемии:
А. Структурные недостатки гемоглобина - наличие гемоглобина М
Б. Неполноценность систем, восстанавливающих метгемоглобин - недостаток метгемоглобинредуктазы, зависящей от восст. НАД (диафораза I)
II Приобретенные причины метгемоглобинемии:
А. Химические причины метгемоглобинемии:
— Нитриты (натрия, амила, этила)
— Нитраты (субнитрат висмута, нитрат аммония)
— Нитробензол
— Нитроглицерин, анилин, фенацетин, ацетиланилид, сульфамиды, сульфаниламид, сульфапиридин, сульфатиазол
Б. Разные причины метгемоглобинемии:
— Кишечная дисфункция (энторегенный цианоз)
— Заражение крови бациллой Clostridium welchii.
В основе наследственных метгемоглобинемий находится генетический механизм. В мировом плане показатель их частоты невелик, при этом не наблюдается расовый характер, хотя отмечается большая частота ферментативной метгемоглобинемий среди эскимосов Аляски, и наличие HbMIwate — среди нескольких семей в Японии. У нас в стране до настоящего времени диагностированы, в одной семье, недостаточность фермента, и в других трех — наличие ГбМ, причем в двух из них определен гемоглобин М вида HbMIwate.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Механизмы развития метгемоглобинемии - патофизиология
Метгемоглобин представляет собой форму окисленного гемоглобина. Последний отличается от нормального гемоглобина наличием в геме железа в состоянии Fe3+, вместо двухвалентного Fe2+, тем самым утрачивая способность обратимо связывать кислород и свойство дыхательного пигмента.
Ежедневно примерно 1% всего гемоглобина преобразовывается в метгемоглобин; в то-же время у лиц в норме, наблюдаются и цифры меньше 2%.
Сохранение метгемоглобина в физиологических пределах и его сокращение в виде функционального гемоглобина (гемоглобин-Fe2+) обеспечиваются ферментативными и иными механизмами защиты эритроцита:
2 гемоглобина — Fe2+ + Н2О2 2 гемоглобина — Fe3+ — ОН (метгемоглобин).
Основной ферментативный механизм образует метгемоглобинредуктазу, зависящую от НАД восстановл. (диафораза I), но существует и второй фермент — метгемоглобинредуктаза, зависящая от восстановленного НАДФ (диафораза II), которая, однако, в физиологических условиях мало активна. Восстановленный глютатион и аскорбиновая кислота участвуют в восстановлении метгемоглобина неферментатувным путем.
В крови метгемоглобин появляется при следующих трех обстоятельствах:
1) его чрезмерном образовании;
2) понижении функции восстановления метгемоглобина в гемоглобин;
3) структурной аномалии глобина с необратимым преобразованием Fe2+ в Fe3+. Метгемоглобин придает крови шоколадно-бурый оттенок, при том синюха развивается когда концентрация достигает примерно 15%, т.е. 2 г/100 мл крови.
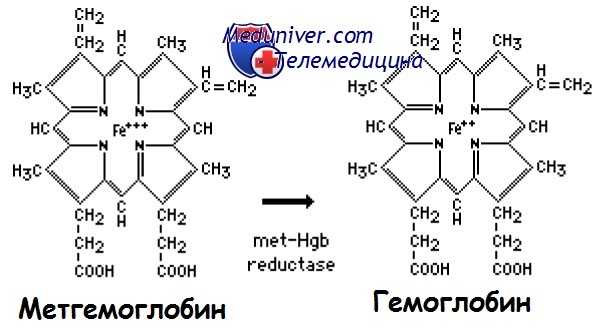
Восстановление метгемоглобина в гемоглобин в норме
Приобретенная метгемоглобинемия наблюдается при чрезмерном образовании метгемоглобина под воздействием отдельных, поступающих в организм веществ. Восстановительный механизм эритроцита действует нормально, однако большая продукция метгемоглобина заглушает его. Развитие и резкость синюхи связаны с количеством и моментом проникновения в организм токсического метгемоглобинизирующего вещества.
На первых месяцах жизни синюха резко выражена даже при отностительно небольшом количестве токсического вещества, по причине более легкой метгемоглобинизации гемоглобина Ф, чем гемоглобина А.
Синюха, развивающаяся при наследственной метгемоглобинемии отличается некоторыми особенностями, которые связаны с механизмом образования метгемоглобина.
Генетический недостаток диафоразы I — основной редуктазы метгемоглобина — заложен в основу энзиматической метгемоглобинемии. Образующееся в организме количество метгемоглобина нормальное, однако оно накопляется по причине недостатка восстановительного фермента. Синюха наблюдается лишь у гомозиготов (рецессивная болезнь), уровень фермента которых нуль или приближается к нулю. Наличие синюхи отмечается непосредственно после рождения.
Тем не менее наличие относительно небольшого количества метгемоглобина (15—30%) даже при полном отсутствии диафоразы I объясняется компенсаторным действием остальных восстановительных механизмов — диафораза II, аскорбиновая кислота, восстановленный глютатион. У гетерозиготов не наблюдается синюха, поскольку невысокий но достаточный уровень фермента обеспечивает восстановление метгемоглобина и нормальную окраску покровов и слизистых оболочек.
Виды гемоглобина М при метгемоглобинемии

У родителей ребенка с метгемоглобинемией ферментативной природы вид нормалный, синюха не обнаруживается.
Отсутствуют доказательства, подтверждающие наличие наследственной метгемоглобинемии за счет недостатка диафоразы II или глютатиона. При наследственной болезни, обусловленной наличием гемоглобина М, образование метгемоглобина объясняется структурной аномалией глобина. Место сдвига находится на уровне связывания гема с глобином, или в непосредственной близости от него, в результате чего налаживаются стойкие связи между аномальной полипептидной цепью и железом гема, с его дальнейшим необратимым фиксированием в виде Fe3+.
Все случаи метгемоглобинемии за счет гемоглобина М представляют собой гетерозиготное состояние в отношение этого аномального гемоглобина. При этой болезни уровень метгемоглобина укладывается в пределы от 15 до 30%, причем объяснение явления следует искать в гетерозиготном состоянии и кодоминантном характере генетической аномалии. Следовательно, в крови подобного больного находится смесь функционального гемоглобина А (70—85%) и гемоглобина М в виде метгемоглобина (15—30%). Гомозиготная форма этого заболевания нежизнеспособна, поскольку все количество гемоглобина в организме оказалось бы нефункциональным — в виде метгемоглобина.
В таких случаях развитие цианоза зависит от структурной аномалии. Так, в случае гемоглобина М Iwate и гемоглобина М Boston, у которых структурня аномалия находится на цепях а, синюха появляется при рождении (цепи альфа включаются и в структуру гемоглобина Ф), в то время как при остальных гемоглобинах М со структурной аномалией на цепях бета, наличие синюхи отмечается на 3—4 месяце жизни, когда зрелый гемоглобин почти полностью заменяет гемоглобин Ф.
Признаки недостатка кислорода отмечаются, в принципе, либо при значительной концентрации метгемоглобина (30—40%), либо при осложнении основного заболевания другим патологическим состоянием, ведущим к уменьшению гемоглобина или его заниженной оксигенации в альвеолах легкого. Следует подчеркнуть, что, у страдающего метгемоглобинемией и показателем гемоглобина 12 г/100 мл крови по существу имеются лишь 7—8 г функционального гемоблобина (60—65%). Хронический тканевой недостаток кислорода объясняет компенсаторную полицитемию, наблюдаемую в отдельных случаях.
Метгемоглобинемия
Метгемоглобинемия – состояние, характеризующееся повышенным содержанием метгемоглобина (окисленного гемоглобина) в крови и тканевой гипоксией. Развитие метгемоглобинемии сопровождается акроцианозом, слабостью, головными болями, головокружением, сердцебиением, одышкой при нагрузке. Характерным признаком метгемоглобинемии служит коричнево-шоколадный цвет крови. Для подтверждения диагноза проводится оценка симптоматики, лабораторные исследования и тесты. При тяжелой степени метгемоглобинемии показана кислородотерапия, введение аскорбиновой кислоты, раствора метиленового синего, в ряде случаев - обменная гемотрансфузия.
МКБ-10
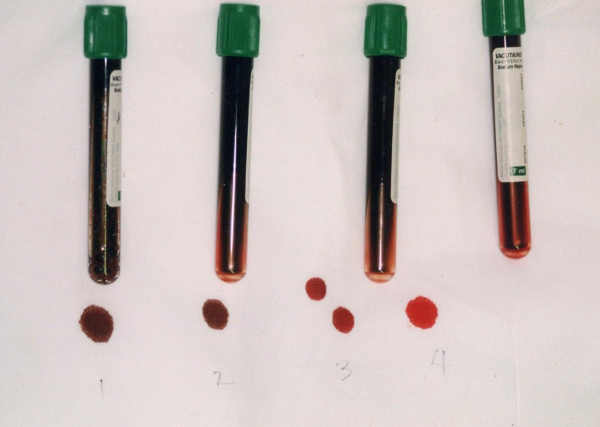
Общие сведения
Метгемоглобинемия – увеличение уровня гемоглобина, содержащего окисленное железо (метгемоглобин – MtHb), в эритроцитах крови. Метгемоглобин относится к так называемым дисгемоглобинам - дериватам гемоглобина, не способным транспортировать кислород. В обычных условиях в крови присутствует небольшое количество метгемоглобина - не более 1% от общего содержания Hb. При метгемоглобинемии эндогенные механизмы оказываются не способными регулировать концентрацию дисгемоглобина, в результате чего страдает кислородно-транспортная функция эритроцитов. В гематологии метгемоглобинемии подразделяются на наследственные и приобретенные. Первые из них распространены среди коренного населения Аляски, Гренландии, Якутии; частота развития приобретенных метгемоглобинемий неизвестна.
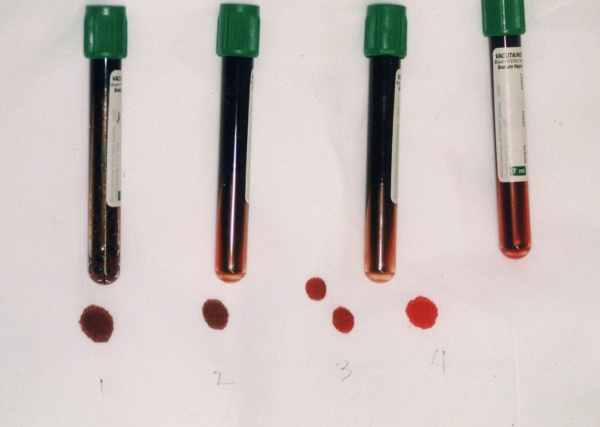
Причины метгемоглобинемии
В процессе метаболических превращений в крови здоровых людей в очень небольших количествах образуются дисгемоглобины: карбоксигемоглобин, сульфгемоглобин, метгемоглобин (0,1-1 %). Вместе с тем, в эритроцитах содержится ряд факторов, которые поддерживают долю фракции метгемоглобина на уровне, не превышающем 1,0-1,5% от общего Hb. В частности, в реакции восстановления метгемоглобина в гемоглобин участвует фермент метгемоглобин-редуктаза. В отличие от оксигемоглобина (НbО2), содержащего восстановленное железо (Fe++), в метгемоглобине содержится окисленное железо (Fe+++), не способное переносить кислород. Поэтому при метгемоглобинемии, прежде всего, страдает кислородно-транспортная функция крови, следствием которой служит тканевая гипоксия.
Наследственные формы метгемоглобинемии представлены либо ферментопатиями (врожденной низкой активностью или отсутствием фермента метгемоглобин-редуктазы), либо М-гемоглобинопатиями (синтезом аномальных белков, содержащих окисленное железо).
В структуре приобретенных (вторичных) метгемоглобинемий выделяют токсические экзогенные и токсические эндогенные формы. Метгемоглобинемии экзогенного происхождения могут быть связаны с передозировкой лекарственных средств (сульфаниламидов, нитритов, викасола, противомалярийных препаратов, лидокаина, новокаина и др.) или отравлением химическими агентами (анилиновыми красителями, нитратом серебра, тринитротолуолом, хлорбензолом, питьевой водой и пищевыми продуктами с высоким содержанием нитратов и др.).
Повышенный уровень MtHb в крови наблюдается у недоношенных и доношенных новорожденных, что связано с низкой активностью фермента метгемоглобин-редуктазы и окислительным стрессом в родах. Однако даже при тяжелой гипоксии и желтухе новорожденных подъем MtHb не столь выражен и клинически значим, чтобы послужить причиной метгемоглобинемии. Однако при диарее, бактериальных и вирусных энтероколитах, в условиях метаболического ацидоза у детей первого года жизни может легко развиться приобретенная эндогенная метгемоглобинемия.
О смешанной форме патологии говорят в том случае, если метгемоглобинемия развивается под воздействием экзогенных факторов у здоровых лиц, являющихся гетерозиготными носителями генов наследственной формы заболевания.
Симптомы метгемоглобинемии
Признаки наследственной метгемоглобинемии становятся заметны в период новорожденности. На коже и видимых слизистых ребенка (в области губ, носогубного треугольника, мочек ушей, ногтевого ложа) заметен цианоз. Кроме наследственной метгемоглобинемии, у детей часто выявляются другие врожденные аномалии - изменения конфигурации черепа, недоразвитие верхних конечностей, атрезия влагалища, талассемия и пр. Нередко дети отстают в психомоторном развитии.
В зависимости от уровня фракции MtHb, выраженность проявлений врожденной и приобретенной метгемоглобинемии может значительно варьировать.
При концентрации MtHb в крови:
Для любых форм метгемоглобинемии характерна грифельно-серая окраска кожных покровов, однако отсутствуют характерные для сердечно-легочных заболеваний изменения ногтевых фаланг по типу «барабанных палочек». Акроцианоз усиливается при охлаждении, употреблении в пищу нитратосодержащих продуктов, при токсикозах беременности у женщин, а также приеме метгемоглобинобразующих медикаментов.
Диагностика метгемоглобинемии
Важным диагностическим признаком метгемоглобинемии служит темно-коричный цвет крови, которая, будучи помещенной в пробирку или на фильтровальную бумагу, не изменяет свой цвет на красный. При положительной пробе проводится спектроскопия, определение концентрации MtHb, активности НАД-зависимой метгемоглобинредуктазы, электрофорез гемоглобина.
В общем анализе крови может присутствовать компенсаторный эритроцитоз, увеличение Hb, ретикулоцитоз, уменьшение СОЭ. При исследовании биохимических показателей крови определяется незначительная билирубинемия, обусловленная увеличением непрямой фракции пигмента. Для хронической метгемоглобинемии типично появление в эритроцитах телец Гейнца-Эрлиха.
У больных с энзимопенической или токсической метгемоглобинемией показательна терапевтическая проба с внутривенным введением метиленового синего – после инъекции цианоз быстро исчезает, а кожа и видимые слизистые приобретают розовую окраску.
При анализе причин метгемоглобинемии важно выяснить, имел ли больной контакт с токсическими веществами, принимал ли метгемоглобинобразующие лекарственные препараты. При подозрении на врожденную метгемоглобинемию изучается родословная, проводится консультация генетика, определяется тип наследования патологии крови. Наследственные метгемоглобинемии требуют дифференциации с врожденными пороками сердца синего типа, аномалиями развития легких и другими состояниями, сопровождающимися гипоксией.
Лечение и профилактика метгемоглобинемии
Больные с отсутствием клинических проявлений не нуждаются в специальной терапии. При значительной концентрации MtHb в крови и развернутой симптоматике метгемоглобинемии назначается медикаментозная терапия, способствующая превращению метгемоглобина в гемоглобин. Такими восстанавливающими свойствами обладают аскорбиновая кислота и метиленовый синий. Аскорбиновая кислота назначается внутрь сначала в больших, а по мере нормализации состояния - в поддерживающих дозах. Раствор метиленовой сини вводится внутривенно. При выраженном цианозе проводится кислородная терапия. Тяжелая форма метгемоглобинемии является показанием к обменному переливанию крови.
Течение наследственной и лекарственной метгемоглобинемии, как правило, доброкачественное. Неблагоприятный исход возможен при тяжелых формах токсической метгемоглобинемии с высоким содержанием MtHb в эритроцитах. Пациентам с подобной патологией следует избегать контакта с метгемоглобинобразующими веществами, переохлаждений и других провоцирующих факторов. Профилактика врожденной метгемоглобинемии заключается в проведении медико-генетической консультации для выявления гетерозиготных носителей среди будущих родителей.
Гемоглобинопатии
Гемоглобинопатии – это группа тяжелых наследственных заболеваний крови, обусловленных нарушением структуры гемоглобина или снижением синтеза одной и более глобиновых цепей. Клиническая картина крайне разнообразна. Общими симптомами являются гемолитическая анемия, увеличение селезенки, поражение костей. Диагностика осуществляется с помощью микроскопии мазка периферической крови, электрофореза гемоглобина, генетических исследований. Для лечения применяют переливание компонентов крови, препараты гидроксимочевины, инфузионную терапию. У тяжелых пациентов выполняется спленэктомия, аллотрасплантация стволовых клеток.
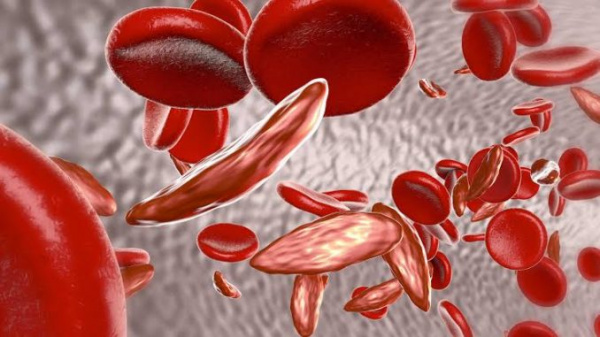
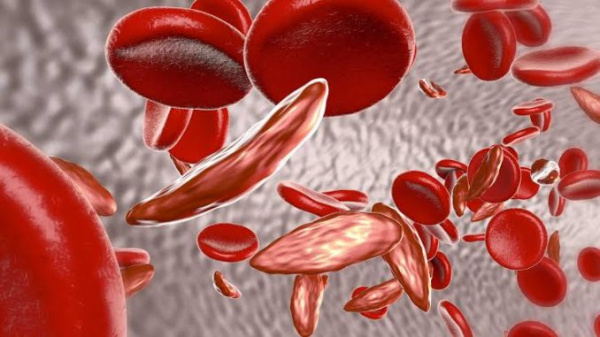
Гемоглобинопатии – ряд врожденных гемолитических анемий, характеризующихся изменением аминокислотной последовательности гемоглобина или подавлением образования цепей глобина. Данные патологии нередко заканчиваются летально уже в раннем детском возрасте. Известно около 50 видов гемоглобинопатий. Самыми частыми и опасными для жизни считаются серповидно-клеточная анемия (СКА), талассемии. Гемоглобинопатии распространены на территории Центральной Африки, Южной Азии и наблюдаются преимущественно у лиц негроидной расы. Талассемии также встречаются в странах Средиземноморья. Ежегодно рождается около 350000 детей с дефектами гемоглобина.
Причины гемоглобинопатий
Гемоглобинопатии относятся к аутосомно-рецессивным генетическим заболеваниям. Качественные гемоглобинопатии развиваются вследствие мутаций генов, ответственных за синтез определенных аминокислот в бета-цепи глобина. В результате происходит замена одной аминокислоты на другую (глутаминовой кислоты на валин, лизин и пр.). Это приводит к образованию аномального гемоглобина, гораздо менее растворимого, чем нормальный гемоглобин А, придающего красным кровяным тельцам иную форму (мишеневидную, серповидную), что нарушает их функции и уменьшает продолжительность жизни.
Количественные гемоглобинопатии обусловлены мутацией генов, которые кодируют целую цепь глобина (чаще альфа и бета). При этом сдвигается баланс между глобиновыми цепями – при недостаточном синтезе альфа-цепей возникает избыток бета-цепей, и наоборот. Уменьшаются размеры эритроцитов, в них снижается содержание гемоглобина, а мембрана становится более подверженной различным повреждениям.
Существуют факторы, провоцирующие тяжелые приступы (кризы). К ним относятся обезвоживание, переохлаждения, инфекции, сопровождающиеся высокой лихорадкой. У женщин обострения нередко развиваются на фоне беременности. Но главный патологический стимул качественных гемоглобинопатий – уменьшение концентрации в крови кислорода (гипоксия). Это может произойти, например, при подъеме на большую высоту (восхождение на гору, полет на самолете), где снижено парциальное давление кислорода воздуха, или при тяжелых болезнях дыхательной системы (пневмонии).
Патогенез
Гемоглобинопатии имеют сходные патогенетические механизмы. Измененная структура гемоглобина предрасполагает к интенсивному гемолизу. Длительно текущая анемия способствует компенсаторной гиперплазии костного мозга. Возникает деформация костей черепа, искривление позвоночника. Развиваются экстрамедуллярные очаги кроветворения, приводящие к увеличению размеров печени и селезенки (гепатоспленомегалии). Вследствие спленомегалии наступает гиперспленизм – усиленная деструкция красных клеток крови синусоидами селезенки.
Из-за регулярного гемолиза эритроцитов печень секретирует в желчь большое количество билирубина, что создает условия для образования камней желчного пузыря. У больных гемоглобинопатиями часто возникает перегрузка железом, как за счет постоянных переливаний крови, так и из-за повышения абсорбции железа желудочно-кишечным трактом. Большое количество железа в тканях усиливает процессы перекисного окисления липидов, что повреждает различные органы.
При качественных гемоглобинопатиях под влиянием сниженного содержания кислорода в крови молекулы нерастворимого аномального гемоглобина растягивают мембрану эритроцитов, что приводит к изменению их формы. Деформированные эритроциты хуже переносят кислород, а также способны приклеиваться к сосудистому эндотелию, тем самым закупоривая мелкие сосуды, вызывая тромбозы, окклюзии и инфаркты.
Классификация
Гемоглобинопатии подразделяются на качественные, обусловленные нарушением структуры (последовательности аминокислот) гемоглобина, и количественные, характеризующиеся снижением образования глобиновых цепей. Качественные гемоглобинопатии представлены следующими формами:
- Серповидно-клеточная анемия (Гемоглобинопатия S). Наиболее частый вид. Подразделяется на гомозиготную форму (собственно СКА) с яркой клинической симптоматикой и гетерозиготное носительство (серповидно-клеточную аномалию), имеющее бессимптомное или легкое течение.
- Гемоглобинопатия С. Клиника схожа с СКА, но менее выражена. Отличается большей степенью спленомегалии, чем при СКА.
- ГемоглобинопатияCS (Африканский ревматизм). По течению напоминает СКА. Преобладают приступообразные костно-суставные боли.
- Гемоглобинопатии Е иD. Протекают с небольшой гемолитической анемией.
- Наследственная метгемоглобинемия. При этой разновидности под влиянием различных факторов образуется окисленная форма гемоглобина – метгемоглобин, который более стойко связывается с кислородом и не отдает его тканям.
- Анемия, вызванная носительством нестабильных гемоглобинов. Это доброкачественная патология, при которой возникает незначительная гемолитическая анемия после приема сульфаниламидных препаратов.
К количественным аномалиям гемоглобина относят:
- Бета-талассемии. Наиболее распространенный вариант. Подразделяется на малую талассемию (гетерозиготное носительство с бессимптомным течением или легкой гемолитической анемией) и большую талассемию (анемию Кули) с развернутой тяжелой клинической картиной.
- Альфа-талассемии. Течение может быть различным в зависимости от количества мутантных генов. В основном сходны с гетерозиготной бета-талассемией.
- Синдром водянки плода c гемоглобином Барт (Hb Bart’s). Наиболее тяжелый вид альфа-талассемии. Ребенок погибает внутриутробно.
- ГемоглобинопатиюH. Благоприятная малосимптомная форма альфа-талассемии.
- Бета-дельта-талассемии. Практически неотличима от бета-талассемии.
- ГемоглобинопатиюLepore. Эта форма развивается вследствие слияния бета-цепей глобина и схожа с бета-талассемией.
Симптомы гемоглобинопатий
Клиническая картина гемоглобинопатий разнообразна. Гетерозиготные больные имеют либо бессимптомное, либо легкое течение. Гомозиготные формы начинают себя проявлять уже с раннего детства (6 месяцев - 1 год). Общая симптоматика включает признаки гемолитической анемии (бледность, желтушность кожи и слизистых, увеличение селезенки), патологию развития скелета – башенный череп (четырехугольный), уплощенная переносица, искривленный позвоночник.
Из-за повышенной секреции билирубина в желчь могут беспокоить симптомы желчнокаменной болезни уже в детском возрасте – тяжесть или ноющие боли в правом подреберье, приступы желчной колики, обесцвечивание кала. На коже голеней часто возникают длительно незаживающие язвы. Для болезней с дефектом в структуре гемоглобина характерно кризовое течение. Самые тяжелые приступы, нередко заканчивающиеся летально, встречаются при серповидно-клеточной анемии.
Вазоокклюзивный криз
Наиболее типичный. Происходит закупорка мелких сосудов различных органов. Дети испытывают боли в длинных трубчатых костях, у них отекают кисти, стопы, что затрудняет движения лучезапястных, голеностопных суставов (hand-foot синдром). Микротромбозы сосудов кишечника вызывают абдоминальные боли. У молодых мужчин нередко развивается приапизм, впоследствии приводящий к эректильной дисфункции. Кризу сопутствует лихорадка, тахикардия, потливость.
Гемолитический криз
При гемолитическом кризе происходит массивное разрушение красных телец с резким снижением содержания гемоглобина, эритроцитов в крови. Усиливается имеющаяся желтушность, бледность кожных покровов, слизистых, возникают лихорадка, поясничные и абдоминальные боли, присоединяются симптомы снижения артериального давления (головокружение, обмороки). Моча приобретает темный цвет за счет большого количества гемоглобина.
Секвестрационный криз
Во время секвестрационного криза также стремительно падает уровень гемоглобина. Но это происходит не из-за гемолиза, а вследствие венозного тромбоза и скопления большого количества крови в расширенных синусоидах печени и селезенке, что и приводит к обеднению общего кровотока. Кроме симптомов тяжелой анемии по причине выраженной гепатоспленомегалии появляются сильные и распирающие боли в левом и правом подреберье.
Апластический криз
Достаточно редкая форма криза. Она развивается при инфицировании парвовирусом В19, который способен угнетать костномозговое кроветворение. Это сопровождается стремительным (но обратимым) снижением концентрации не только эритроцитов, но и всех других клеток периферической крови (лейкоцитов, тромбоцитов). Поэтому к признакам анемии присоединяется геморрагический синдром (кровотечения из носа, десен), различные инфекции (в основном ОРВИ).
Осложнения
Общими осложнениями гемоглобинопатий считаются ЖКБ, патологические переломы длинных трубчатых костей. Гетерозиготные формы редко сопровождаются неблагоприятными событиями, так как имеют легкое течение. При количественных гемоглобинопатиях из-за отложения избыточного количества железа во внутренних органах развивается сердечная недостаточность, цирроз печени, сахарный диабет 2 типа.
Качественные патологии гемоглобина характеризуются широким спектром неблагоприятных последствий. Наиболее опасными считаются эмболия легочных сосудов, инфаркт миокарда, ОНМК, которые примерно у 10% пациентов приводят к смерти. Закупорка микрососудов, питающих кости, ведет к асептическому некрозу головки бедренной кости (АНГБК). Вследствие постоянных инфарктов селезенки возникает функциональный асплезнизм, из-за чего часто развиваются бактериальные инфекции (бронхиты, пневмонии) с тяжёлым течением, нередко с летальным исходом.
Диагностика
Курацией больных с гемоглобинопатиями занимаются врачи-гематологи, генетики. Во время общего осмотра обращается внимание на цвет кожных покровов (бледность, желтуха), конституциональные нарушения (задержку нервно-психического, физического развития ребенка, аномалии строения скелета). Дополнительное обследование включает:
Гемоглобинопатии дифференцируют с другими врожденными гемолитическими анемиями (мембранопатиями, ферментопатиями, микросфероцитарной анемией Минковского-Шоффара). Постоянные тромбозы нужно дифференцировать от различных тромбофилий. Перегрузку железом следует отличать от наследственного гемохроматоза. Анемия, оссалгии требуют исключения злокачественных миелопролиферативных заболеваний.
Лечение гемоглобинопатий
Людям, страдающим гемоглобинопатиями, требуется проведение сложной многокомпонентной терапии, поэтому все пациенты с гомозиготными формами подлежат обязательной госпитализации в гематологический стационар. Больным с гетерозиготными формами лечение не показано. Основные принципы ведения качественных и количественных патологий гемоглобина несколько отличаются друг от друга.
Консервативная терапия
Подбор консервативной терапии производится с учетом вида гемоглобинопатии, течения заболевания, наличия тех или иных осложнений. Оценивается как клиническая симптоматика, так и лабораторные данные (главным образом, показатели красной крови). Выделяют следующие направления лечения:
- Купирование кризов. При вазоокклюзивных кризах используются обезболивающие препараты (нестероидные противовоспалительные средства), а при выраженном болевом синдроме - наркотические анальгетики. Также для подавления преципитации деформированных эритроцитов назначаются ингаляции кислородом, пероральная или внутривенная регидратация.
- Предупреждение кризов. Для постоянного приема больным качественными гемоглобинопатиями назначается гидроксимочевина. Этот препарат стимулирует образование фетального гемоглобина, подавляющего экспрессию гена, ответственного за синтез аномальных нерастворимых гемоглобинов, что уменьшает склонность эритроцитов к деформации, снижает частоту кризов.
- Терапия осложнений. Инфекционные осложнения до получения результатов бактериологических исследований лечат антибактериальными препаратами, активными против пневмококка, гемофильной палочки, менингококка. Используются антибиотики из группы пенициллинов (амоксициллин). При развитии тромбозов применяются антикоагулянты (низкомолекулярные, нефракционированные гепарины).
- Гемотрансфузии. Так как анемия у пациентов с количественными патологиями гемоглобина всегда тяжелая, основу лечения составляют регулярные гемотрансфузии. Людям, страдающим качественными гемоглобинопатиями, переливания крови проводятся только при секвестрационных, гемолитических, а также апластических кризах.
- Борьба с перегрузкой железа и дефицитом фолатов. Для выведения избытка железа из организма используются хелатирующие средства (дефероксамин). Этот препарат обычно назначается вместе с аскорбиновой кислотой, так как она потенцирует хелатирующее действие дефероксамина. Из-за постоянного гемолиза у больных повышен расход фолатов, поэтому им показан длительный прием больших доз фолиевой кислоты.
Хирургическое лечение
Для ряда пациентов с выраженными признаками гемолиза эффективным лечением является спленэктомия – оперативное удаление селезенки. Другой хирургический вид лечения, позволяющий добиться полной ремиссии – аллотрансплантация гемопоэтических стволовых клеток. Однако этот метод применяется редко, только в очень тяжелых случаях, так как он сопряжен с высокой частотой летальных исходов. При холелитиазе выполняется холецистэктомия.
Экспериментальное лечение
В настоящее время ведутся клинические исследования по поиску лекарства для излечения гемоглобинопатий. Есть успешные результаты генной терапии СКА. Суть лечения заключается во введении в стволовые клетки пациента гена, кодирующего синтез нормальной бета-глобиновой цепи, с помощью обезвреженного лентивируса. Препарат называется LentiGlobin BB305. Его применение привело к улучшению показателей крови, что позволило отказаться от постоянной стандартной терапии. Также проводятся испытания данного препарата при бета-талассемии.
Прогноз и профилактика
Гемоглобинопатии являются тяжелыми заболеваниями с опасными жизнеугрожающими осложнениями. Пациенты с гомозиготной альфа-талассемией умирают еще до рождения в утробе матери. Больные бета-талассемией погибают до наступления пубертатного возраста от сердечной недостаточности. Люди, имеющие качественные гемоглобинопатии, при грамотном лечении могут прожить дольше 50 лет. Основная причина смерти – бактериальные инфекции, тромботические осложнения. При гетерозиготных формах заболевания в большинстве случаев продолжительность жизни не отличается от общей популяции.
Первичная профилактика проводится среди семей, имеющих высокий риск развития гемоглобинопатий. Она заключается в пренатальной диагностике с прерыванием беременности по медпоказаниям. Пациенты, страдающие качественными аномалиями гемоглобина, обязательно должны получить вакцину от гемофильной палочки, пневмококка, менингококка. Детям от 4 месяцев до 6 лет показано длительное применение пенициллиновых антибиотиков для профилактики инфекций. То же касается больных, перенесших спленэктомию.
Гемолитическая анемия
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
Гемолитическая анемия (ГА) - малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
Причины
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Центральным звеном патогенеза ГА является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. При аутоиммунном механизме анемии происходит образование антиэритроцитарных АТ (тепловых, холодовых), которые вызывают ферментативный лизис мембраны эритроцитов. Токсические вещества, являясь сильнейшими окислителями, разрушают эритроцит за счет развития метаболических, функциональных и морфологических изменений оболочки и стромы красных кровяных телец. Механические факторы оказывают прямое воздействие на клеточную мембрану. Под влиянием этих механизмов из эритроцитов выходят ионы калия и фосфора, а внутрь поступают ионы натрия. Клетка разбухает, при критическом увеличении ее объема наступает гемолиз. Распад эритроцитов сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии- анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) – обусловлены воздействием антител
- токсические – анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические - анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Симптомы
Наследственные мембранопатии, ферментопении и гемоглобинопатии
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом - обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов – еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические анемии связаны с недостатком определенных ферментов эритроцитов (чаще - Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет. Особенностям клинического течения гемоглобинопатий - талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Приобретенные гемолитические анемии
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия. Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ – при микросфероцитозе, печеночная недостаточность – при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Лечение
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов.
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
4. Клинические рекомендации по диагностике и лечению аутоиммунных гемолитический анемий/ Цветаева Н.В., Никулина О.Ф. - 2014.
Читайте также:
- Дисфункция головного мозга в зависимости от локализации
- Интраперикардиальная перевязка сосудов. Эффективность трансперикардиального доступа
- Венозный отток от тонкой кишки. Лимфатические сосуды тонкой кишки. Лимфатические узлы тонкой кишки. Иннервация тонкой кишки.
- Лучевая анатомия (КТ, МРТ анатомия) диафрагмы
- Пересадка ядра при генетических болезнях. Терапевтическое клонирование