Миеломенингоцеле: атлас фотографий
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Фиксированный спинной мозг со спина бифида: менингоцеле, миеломенингоцеле или Tethered Cord Syndrome
Последнее редактирование: 17/01/2020, Доктор Мигель Б. Ройо Сальвадор, Номер в коллегии: 10389. Нейрохирурга и Невролога.
Определение
В норме спинной мозг находится внутри позвоночного канала и фиксируется только зубовидными связками и концевой нитью, связкой, соединяющей кончик спинного мозга с первыми позвонками копчика.
Когда подвижность спинного мозга внутри позвоночного канала ограничена и зафиксирована присутствием спина бифида, которую заметно внешне, такая мальформация носит название Синдрома фиксированного спинного мозга со спина бифида или “Tethered Cord Syndrome”.
Спина бифида – это мальформация, возникающая при неполном развитии нервной трубки (впоследствии превращается в головной и спинной мозг плода), при которой существует отверстие в позвонках.
При спина бифида с менингоцеле мягкие оболочки, окружающие спинной мозг, выпячиваются через костный дефект позвоночника и формируют мешок с жидкостью. (Рис.1).
При “открытой” спина бифида с миеломенингоцеле также существует отверстие в нескольких позвонках в средней или нижней части позвоночника (Рис.1). Но в этом случае, выпячивание включает в себя мягкие оболочки и ткани спинного мозга, а также корешки спинальных нервов. Грыжа часто бывает открытой, что ставит под угрозу жизнь ребенка.
При синдроме фиксированного спинного мозга со спина бифида происходит механическое натяжение всей нервной системы, костных структур черепа и позвоночника, что приводит к образованию синдрома Арнольда Киари II типа, вторичных сколиоза и сирингомиелии.
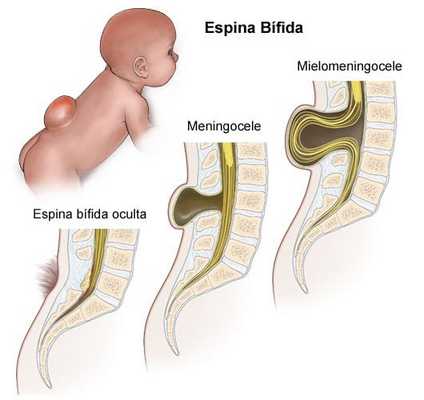
Рис.1 Типы спина бифида (снизу вверх): Скрытая спина бифида, Менингоцеле, Миеломенингоцеле.
Диагностика
Диагноз фиксированного спинного мозга со спина бифида может быть поставлен в пренатальный период. Во время беременности проводятся различные контрольные исследования для обнаружения возможных врожденных дефектов. Для обнаружения спина бифида проводится:
- Альфафетопротеин в плазме крови
- Проба на подтверждение высокого уровня альфафетопротеина
- Другие дополнительные анализы крови
Симптомы
Симптомы при спина бифида зависят от размера и расположения мальформации, покрыта ли она кожей или нет, видны ли нервы, в какой степени повреждена нервная структура и нервы.
Мальформация может повлиять на три основные системы организма: центральную нервную систему, опорно-двигательную систему и мочеполовую систему. Может спровоцировать разные степени паралича и потерю чувствительности в нижних конечностях, а также различные осложнения в функциях кишечника и мочеполовой системы. Обычно повреждены все нервы, расположенные ниже уровня мальформации. Поэтому, чем выше мальформация, тем больше повреждений нервов и потеря двигательных функций и чувствительности.
Причины
Обычно нервная трубка плода формируется на первых этапах развития беременности и закрывается примерно на 28 день после зачатия. У младенцев со спина бифида часть нервной трубки не закрывается или не формируется, что приводит к дефектам на уровне спинного мозга и позвоночника.
Спина бифида имеет наследственный фактор, но не всегда. Пока неизвестны все элементы и механизмы, приводящие к нарушениям в закрытии нервной трубки. Речь идет о нарушении, происходящем на этапе развития индивида, и часто оно связано с низким уровнем фолиевой кислоты во время беременности.
Факторы риска
Факторы, которые влияют на формирование спина бифида с менингоцеле или миеломенингоцеле у плода:
- Наследственность, случаи с проблемами закрытия нервной трубки в семье.
- Недостаток фолиевой кислоты.
- Диабет.
- Ожирение.
- Повышенная температура тела материво время беременности.
- Прием антиконвульсантов и других препаратов во время беременности.
- Иные причины окружающей среды во время беременности.
Осложнения
Первым осложнением спина бифида с менингоцеле или миеломенингоцеле является фиксированный спинной мозг. Это мешает правильному развитию плода.
Позднее, к этому могут добавиться в разных сочетаниях следующие заболевания:
Другие возможные осложнения:
- Проблемы с передвижением и походкой
- Деформация стоп
- Другие ортопедические осложнения
- У детей с миеломенингоцеле и гидроцефалией могут быть проблемы с обучением.
- По мере роста у детей со спина бифида могут наблюдаться дополнительные проблемы, такие как аллергия на латекс, кожные заболевания, желудочнокишечные расстройства и депрессия.
Лечение
Существует несколько методов лечения фиксированного спинного мозга со спина бифида, в зависимости от момента его обнаружения, типа и связанных с ним заболеваний:
- Внутриутробное хирургическое вмешательство: открывается и восстанавливается позвоночный канал плода, лечение проходит до 26 недели беременности в специализированных центрах пренатальной хирургии, с интенсивным уходом, с командами педиатров. Помогает снизить риск преждевременных родов и избежать последующих осложнений заболевания: инфекций и гидроцефалии.
- Хирургическое закрытие дефекта после рождения: цель операции – избежать вторичного инфицирования нервных тканей и любой другой травмы открытой зоны спинного мозга. В случае менингоцеле мягкая оболочка мозга возвращается на место и закрывается проход между позвонками. В случае миеломенингоцеле существует риск повреждения нервных окончаний, для избежания инфекции открытых нервов для защиты сверху ставится заплатка из мышц и кожи. Иногда также устанавливается дренаж для контроля гидроцефалии.
- Лечение последствий: младенцам с миеломенингоцеле может потребоваться больше операций для лечения последствий. Кроме того, этим пациентам в течение их развития необходимы дополнительные курсы лечения для контроля за кишечником и мочевым пузырем, для ходьбы и передвижения, чтобы улучшить качество их жизни.
Список литературы:
- Cahit Kural, Ilker Solmaz, Ozkan Tehli, Caglar Temiz, Murat Kutlay, Mehmet K. Daneyemez, Yusuf Izci. Evaluation and Management of Lumbosacral Myelomeningoceles in Children Eurasian J Med 2015; 47: 174-8
- Kenga Sivarajah, Sophie Relph, Radha Sabaratnam, Spyros Bakalis. Spina bifida in pregnancy: A review of the evidence for preconception, antenatal, intrapartum and postpartum care. Obstet Med. 2019 Mar; 12(1): 14–21. Published online 2018 May 17.
- Gutiérrez-Cabrera J., Pedroza-Ríos K. G., Cuéllar-Martínez S. Médula anclada en pacientes pediátricos y adolescentes. Revisión de 16 casos. Revista médica del Hospital General de México, S. S., Vol. 70, Núm. 2 Abr.-Jun. 2007 pp. 62-66.
- Lichtenstein B.W. Spinal dysraphism. Spina Bifida and myelodysplasia. Arch Neurol Psychiatry 44:792-809, 1940.
- Shokei Yamada. “Tethered cord Syndrome” American Association of Neurological Surgeons Publications Commitee 1996. ISBN 1-879284-37-5.
Для того, чтобы понять разницу между диагнозами Фиксированный спинной мозг со спина бифида, Фиксированный спинной мозг со скрытой спина бифида и Заболевание концевой нити, читайте статьи по ссылкам или смотрите видео).
Свяжитесь с нами

Меня зовут Нина, я буду ассистентом в Вашей консультации.
Все консультации, полученные через этот формуляр или по электронной почте Барселонского Института Киари & Сирингомиелии & Сколиоза, передаются медицинскому отделу, и ответы проверяются доктором М. Б. Ройо Сальвадор.
Менингомиелоцеле
Менингомиелоцеле – это грыжа спинномозгового канала, при которой происходит выпячивание тканей и вещества спинного мозга через костный дефект позвоночного столба. Клиническая картина включает в себя наличие грыжеобразного выпячивания на спине ребенка в поясничной или крестцовой области. Сразу или с возрастом возникает нарушение иннервации нижерасположенных сегментов, вследствие чего развиваются тазовая дисфункция, парапарезы или параплегия. Диагностика основывается на наружном осмотре, подтверждении поражения ЦНС при помощи КТ и МРТ. Лечение хирургическое с последующей симптоматической терапией.
Общие сведения
Менингомиелоцеле или spina bifida cystica – это одна из форм спинального дизрафизма, которая проявляется выходом тканей спинного мозга за пределы позвоночного канала через костный дефект дужек. Впервые патология была описана в 1641 году Н. Тульпиусом. Распространенность составляет 5-20 на 10000 новорожденных. Является тяжелым заболеванием, которое вызывает серьезные неврологические нарушения и в некоторых случаях приводит к полному обездвиживанию больного. При менингомиелоцеле грыжевой мешок содержит оболочки спинного мозга, спинномозговую жидкость и корешки спинальных нервов. Чаще заболевание наблюдается при беременности матери в возрасте после 35 лет. В 6-8% случаев прослеживается наследственная склонность, что свидетельствует о генетическом характере патологии. При адекватном своевременном лечении прогноз относительно благоприятный.
Причины менингомиелоцеле
Менингомиелоцеле развивается при наличии одного или нескольких факторов риска со стороны матери: прием фармакологических средств (оральные контрацептивы, препараты из групп салицилатов, вальпроатов, амфетаминов); употребление алкогольных напитков, табачных изделий, наркотиков, других тератогенных веществ; недостаточность микроэлементов и питательных веществ (в частности – Zn, Fe, фолиевая кислота) в рационе беременной; TORCH-инфекции (чаще всего – вирусы краснухи, гриппа и парагриппа). Также патология может иметь наследственный характер – установлен аутосомно-рецессивный механизм передачи заболевания, однако в 90-95% случаев семейный анамнез не отягощен.
Нервная трубка образуется из нервной пластинки на 19-20 день гестации. В нормальных условиях ее анатомическое закрытие происходит на 22-24 сутки, в результате чего остаются только верхнее и нижнее отверстие. Существует закономерность: чем позднее развивается менингомиелоцеле – тем ниже его локализация. Патогенез менингомиелоцеле досконально не изучен. На данный момент в педиатрии существуют 3 основных теории, объясняющих механизм развития данной патологии. Первая – это патология закрытия нервной трубки или теория Реклингхаузена. Она объясняет развитие спинального дизрафизма дефектом нейроэктодермы, возникающим в периоде раннего онтогенеза – на 25-30 день после оплодотворения.
Вторая – гидродинамическая или теория Моргани. Ее суть заключается в повышении давления внутри спинномозгового канала в I триместре беременности. Повышение давления провоцирует выпирание оболочек с последующим формированием костного дефекта и грыжи. Косвенным подтверждением данной теории является сопутствующая гидроцефалия в 90% случаев заболевания. Третья теория объясняет развитие менингомиелоцеле нарушением темпа роста тканей спинномозгового канала. При этом скорость дифференциации костных тканей позвоночного столба отстает или опережает развитие нервной трубки, из-за чего формируется дефект позвоночного столба.
Симптомы менингомиелоцеле
Клинические проявления менингомиелоцеле наблюдается уже с момента рождения ребенка. Основной признак – наличие характерного «мешка», который являет собой грыжеобразное выпячивание спинного мозга на спине ребенка. В некоторых случаях данное образование покрыто тонким шаром эпидермиса, но зачастую в дефекте позвоночного столба визуализируются непосредственно оболочки спинномозгового канала и нервные корешки. Наиболее характерная локализация – поясничный или крестцовый отдел. Примерно у половины новорожденных с изолированной формой изменения общего состояния изначально не возникает, однако с возрастом у всех детей появляются неврологические нарушения. Степень поражения напрямую зависит от уровня спинного мозга, на котором сформировалась грыжа.
При развитии менингомиелоцеле ниже 4 поперечного сегмента (L4) наблюдаются нарушения иннервации мочевого пузыря, реже – терминальных отделов кишечного тракта. Типичное проявление – недержание мочи. В более тяжелых случаях помимо тазовой дисфункции развиваются расстройства чувствительности и моторной функции нижних конечностей по типу парапареза. При локализации грыжевого выпячивания выше уровня 3 поперечного сегмента (L3) возникает полная параплегия, приводящая к тотальному обездвиживанию ног. Менингомиелоцеле зачастую сочетается с гидроцефалией и мальформацией Арнольда-Киари II типа с характерными для них клиническими проявлениями: увеличением размеров головы, рвотой, бессонницей, конвульсиями, атаксией, нарушением акта глотания, головными болями в области затылка, задержкой психофизического развития и т. д. Значительно реже в виде осложнения возникает эпилепсия.
Начиная с 12-ти месячного возраста, часто наблюдаются задержка в физическом развитии, набор излишней массы тела и деформации нижних конечностей. Ожирение в основном связано с малоподвижным или неподвижным образом жизни, а деформация – с отсутствием осевой нагрузки на ноги и отсутствием сопротивления для внутренних групп мышц стопы. На фоне дисфункции мочевого пузыря учащается пузырно-мочеточниковый рефлюкс, увеличивается вероятность развития инфекционных заболеваний мочевой системы, что может привести к почечной недостаточности.
Диагностика менингомиелоцеле
Диагностика менингомиелоцеле базируется на сборе анамнестических данных, проведении физикального обследования, использовании лабораторных и инструментальных методов исследования. При сборе анамнеза педиатр может установить факторы риска заболевания или возможную этиологию. Физикальное исследование заключается в непосредственном осмотре грыжевого выпячивания, определении уровня поражения и выявлении симптомов сопутствующих патологий. В общих лабораторных тестах (ОАК, ОАМ) при изолированной форме менингомиелоцеле отклонения от нормы не выявляются. При наличии в анамнезе данных, указывающих на возможную этиологию, могут использоваться специфические анализы – определение концентрации фолиевой кислоты, железа, цинка в плазме крови; ПЦР или ИФА на возбудителей TORCH-инфекций. Для уточнения уровня поражения спинного мозга и размера дефекта применяются методы нейровизуализации – компьютерная и магнито-резонансная томография. Также данные исследования позволяют выявить сопутствующие аномалии строения ЦНС – гидроцефалию, мальформацию Арнольда-Киари и другие. Дифференциальная диагностика менингомиелоцеле проводится с другой формой расщепления позвоночника – менингоцеле.
Лечение менингомиелоцеле
Основное лечение менингомиелоцеле осуществляется хирургическим путем. Суть – послойное закрытие дефекта и формирование спинномозгового столба. Ход операции: выделение и вскрытие грыжевого мешка, погружение тканей ЦНС в позвоночный канал, удаление дефекта и сшивание его остатков, коррекция дужек при помощи миофасциального лоскута. После операции, несмотря на восстановление нормальной структуры спинного мозга, избежать неврологических нарушений удается редко, т. к. ткани спинного мозга и корешков во время внутриутробного развития подвергаются необратимой дегенерации. При сопутствующей гидроцефалии также проводится ее нейрохирургическая коррекция.
Симптоматическая терапия подразумевает лечение развившихся осложнений. Назначают уросептики при частых инфекциях мочеполовой системы, оксибутинина гидрохлорид при дисфункции мочеиспускания и антихолинергические средства для стимуляции нейромедиаторной передачи. Важная роль отводится коррекции рациона ребенка, направленной на компенсацию дефицита микроэлементов. Применяют препараты цинка и железа, фолиевой кислоты, витамина С и В12. При частых запорах рекомендовано увеличить объем потребляемой жидкости.
Прогноз и профилактика менингомиелоцеле
Прогноз при менингомиелоцеле зависит от эффективности проводимого лечения. Как правило, при своевременной хирургической коррекции патологии, адекватной симптоматической терапии и рациональном питании исход достаточно благоприятный.
Специфической профилактики для данной патологии не существует. При отягощенном семейном анамнезе беременной проводится амниоцентез с целью антенатальной диагностики дефектов строения позвоночного столба и спинного мозга плода. Кроме того, используются общепринятые методы исследования в период беременности: УЗИ, определение концентрации альфа-фетопротеина в околоплодных водах. При выявлении TORCH-инфекций осуществляется их лечение и полноценное обследование матери и плода после проведенной терапии. Беременным рекомендуется увеличить количества метионина, витамина В12 и фолиевой кислоты в рационе, т. к. данные вещества снижают риск расщепления позвоночника ребенка.
Аномалия Киари ( Синдром Арнольда-Киари )
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
МКБ-10
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
- Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
- Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
- Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
- Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Болезнь шарко мари тута: причины, 8 симптомов, 3 метода лечения
Шарко — Мари — Тута болезнь (J. М. Charcot, 1825—1893, французский невропатолог; P. Marie, 1853—1940, французский невропатолог; Н. Н. Tooth, 1856—1925, английский невропатолог) — см. Амиотрофия наследственная невральная.
Выделяют формы заболевания с учетом характера повреждения нервных окончаний, принадлежащих периферическому отделу нервной системы. Патология 1 типа развивается вследствие демиелинизации – разрушения миелиновой оболочки, покрывающей нервы. В норме миелиновая оболочка улучшает передачу нервных импульсов. Дебютирует чаще в среднем (4-5 лет) детском возрасте, реже в подростковом периоде.
Первые симптомы: слабость в стопах, атрофические изменения в мышцах дистальных отделов ног. Атрофические изменения в мышечной ткани кистей рук развиваются позже. Вместе с утратой чувствительности (неспособность различать температуру, воспринимать болевые раздражители, вибрацию) в зоне кистей рук и стоп ног наблюдается понижение (иногда выпадение) сухожильных рефлексов.
Полинейропатия Шарко-Мари-Тута 2 типа развивается на фоне повреждения аксонов – ответвлений нейронов, посредством которых осуществляется связь между отдельными элементами нейрональной сети. Это медленно прогрессирующая форма. Дебютные симптомы могут появиться у детей старшего возраста. В ходе инструментальной диагностики выявляются относительно нормальные показатели скорости проведения нервных импульсов.
Параллельно наблюдается уменьшение разницы между амплитудными значениями потенциалов действия (волна возбуждения) чувствительных нервных клеток. Другая особенность – полифазные (многофазные) потенциалы действия в области мышечных волокон. Биопсия показывает аксональную дегенерацию валлеровского типа – процесс разрушения участка аксона.
Медицинская библиотека
- Акушерство и гинекология
- Анатомия и физиология
- Анестезиология и реаниматология
- Болезни органов дыхания
- Болезни пищеварительной системы
- Болезни сердечно-сосудистой системы
- Гематология
- Дерматология и венерология
- Инфекционные болезни
- Информация
- Контрацепция
- Красота, здоровье, долголетие
- Неврология и невропатология
- Онкология
- Оториноларингология
- Офтальмология
- Педиатрия
- Психология и психиатрия
- Сексология и сексопатология
- Стоматология
- Судебная медицина
- Терапия
- Урология и нефрология
- Фармакология
- Хирургия
- Эндокринология
- Энциклопедия
- Медицинские термины
- Ш
Перонеальная амиотрофия (болезнь Шарко … Перонеальная (невральная) амиотрофия … Болезнь Келера II: атлас фотографий
Основные формы болезни Шарко — Мари — Тута
| Этот раздел не поможете проекту, исправив и дополнив его следующей информацией: о формах болезни Шарко — Мари — Тута. |
Существуют различные формы болезни Шарко — Мари — Тута. Основные формы имеют обозначения ШМТ1, ШМТ2, ШМТ3, ШМТН4, ШМТ5, ШМТ6, ШМТ-ДП, ШМТ-РП и ШМТХ[3].
Причиной ШМТ1 является нарушение миелиновой оболочки периферических нервов, эта форма называется миелинопатия и имеет несколько типов со сходными симптомами. Первые признаки болезни появляются, как правило, в подростковом возрасте. Пациенты испытывают мышечную слабость в ногах, у них происходит атрофия мышц дистальных отделов нижних конечностей, где позднее слабеет и утрачивается чувствительность. Скорость проведения импульса по срединному нерву снижена и составляет менее 38 м/с. У пациентов выявляется сегментарная демиелинизация и ремиелинизация. При биопсии нервных волокон выявляется гиперплазия шванновских клеток с формированием характерного морфологического признака «луковичные головки».
Болезнь Шарко Мари Тута: причины, симптомов, метода лечения
Шарко Мари Тутанаследственных невропатий 4 варианта недугаСиндром Шарко первого (или демиелинизирующего) типа наблюдается у 60% пациентов.Второй вариант патологии, именуемый также аксональным и встречающийся в 20% случаевТретьим вариантом патологии является тот, который наследуется через Х хромосому доминантно.
- Невральная амиотрофия четвёртого типа считается демиелинизирующей и наследуется аутосомно-рецессивно, то есть существует шанс рождения здоровых детей у больных родителей.
- Основной и единственной причиной любого типа болезни Шарко-Мари-Тута являются точечные мутации в генах.
- Демиелинизирующий тип патологии также связывают с приобретенной аутоиммунной агрессией, при которой клетки, создающие наружную оболочку нерва, воспринимаются организмом как чужеродные и подвергаются уничтожению.
- Существуют также факторы риска, способствующие более яркому проявлению симптомов болезни Шарко-Мари-Тута и усугублению уже имеющейся клинической картины.
- К триггерам недуга относят:
- употребление алкоголя;
- применение потенциально нейротоксичных препаратов. К примеру, винкристина, солей лития, метронидазола, нитрофуранов и т.п.
Общие симптомы
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Первый тип
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.
синдром Руси-Леви
Второй тип
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
Что нужно для подтверждения диагноза
- демиелинизация и ремиелинизация волокон с формированием «луковичных головок»;
- снижение доли крупных миелинизированных волокон;
- атрофия аксонов с уменьшением их диаметра.
Подходы к лечению болезни Шарко
Хирургическое лечение
Физиотерапевтические методы
Основные группы лекарственных препаратов
Прогноз
Усугубить состояние больного могут беременность, а также использование запрещённых при этой патологии препаратов: Тиопентала, Винкристина и др. В 20-25% случаев ухудшение связывают с присоединением аутоиммунного процесса, при нём клетки, целью которых является защита организма от чужеродных антигенов, начинают атаковать миелиновую оболочку собственных нервов.
Осложнения болезни Шарко
Заключение
Дифференциация невральной амиотрофии
Невральную амиотрофию иногда трудно дифференцировать от различных хронических полиневритов, при которых также наблюдаются дистальные атрофии мышц. В ее пользу говорят наследственный характер и прогрессирующее течение болезни. От дистальной миопатии Гоффманна невральная амиотрофия отличается фасцикулярными подергиваниями в мышцах, нарушениями чувствительности, отсутствием поражения мышц туловища и проксимальных отделов конечностей, а также электромиографической картиной.
Болезнь шарко мари тута — проявление … Рассекающий остеохондрит (РО): атлас … Болезнь шарко-мари-тута: наследственное …
Гипертрофический интерстициальный неврит Дежерина — Сотта отличается от невральной амиотрофии значительным утолщением (часто узелковым) нервных стволов, атаксией, сколиозом, более грубыми изменениями болевой чувствительности, частым присутствием зрачковых нарушений, нистагмом.
Важно! Регистры пациентов с болезнью Шарко-Мари-Тута
- Международный регистр (реестр) пациентов с БШМТ -clinical-trial-registry/
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку болезнь Шарко-Мари-Тута является редким (орфанным) заболеванием.
Для этого в разных странах ведутся реестры пациентов с БШМТ — базы данных по генетической и клинической информации о людях, страдающих БШМТ и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных данным заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Найден способ лечения болезни Шарко Мари Тута
- Болезнь Шарко Мари Тута — неизлечимое заболевание, наследственная болезнь периферической нервной системы, объединяющая в себе невропатии двух типов — I и II.
- Группа исследователей, занимающихся научными исследованиями в Антверпенском университете, сообщили, что они смогли найти лекарство от этой болезни.
- Болезнью Шарко Мари Тута страдает 1 человек на 2500 особей, что делает данное заболевание наиболее распространенным среди всех болезней периферической нервной системы, передающихся по наследству.
ШМТ характеризуется потерей мышечной ткани из-за денервации и сенсорных нарушений (преимущественно в стопах и голенях, но и в руках на поздних стадиях заболевания). В настоящее время не представляется возможным вылечить или предотвратить ШМТ, которая влияет как на детей, так и на взрослых.
Исследования в области молекулярно-биологических процессов, ведущих к болезни Шарко Мари Тута важны, потому что они способствуют развитию диагностики и предлагают возможные методы лечения.
Ранее исследователи отмечали, что ряд пациентов с данным заболеванием имеют мутации в гене HSPB1, который находится в 7-й хромосоме, кодирующем примерно 27 кДа белка B1.
Миеломенингоцеле: атлас фотографий Миеломенингоцеле: атлас фотографий ЛЕКЦИИ ПО МЕДИЦИНСКОЙ ГЕНЕТИКЕ — PDF …
До сих пор было неизвестно, как влияют мутации в гене HSPB1 на проблемы периферической нервной системы, однако ученые знали, что подобные изменения приводят к дегенерации нервных пучков и мышечной слабости.
Для исследования процесса ученые выделили мутировавший ген у больных пациентов, чтобы внедрить его в нейроны мышей. Мыши продемонстрировали все симптомы, в том числе и моторные, а также атрофию мышц и слабость, деформацию стоп и перонеальную походку — животные продемонстрировали весь список признаков заболевания, наблюдаемый у человека.
Исследуя развитие болезни у мышей, ученые обнаружили, что транспортировка митохондрий (клеточных электростанций) к аксонам сильно нарушена, что, вероятно, обусловлено повреждением микротрубочек, по которым транспортируются митохондрии. Это может привести к хроническому недостатку митохондрий в нервных окончаниях, вызывая вырождение клеток.
В качестве варианта лечения ученые рассматривают возможность использования ингибиторов HDAC6, катализирующих восстановительные процессы белков, из которых строятся микротрубочки. Опыты на мышах показали, что использование данных ферментов не только способно остановить развитие болезни, но и обратить симптомы вспять — восстановить мышцы, например.
Формы болезни Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута является результатом аномалий в аксоне периферической нервной клетки, а не в оболочке миелина. Он менее распространен, чем CMT1. CMT2A, наиболее распространенная аксоновская форма CMT, вызвана мутациями в Mitofusin 2, белке, связанном с митохондриальным слиянием. CMT2A также был связан с мутациями в гене, который кодирует белок 1B-бета-члена семейства кинезина, но в других случаях он не реплицируется.
Кинезины — это белки, которые действуют как двигатели, чтобы помочь обеспечить транспортировку материалов вдоль клетки. Другие менее распространенные формы CMT2 были недавно идентифицированы и связаны с различными генами: CMT2B (связанный с RAB7), CMT2D (GARS). CMT2E (NEFL), CMT2H (HSP27) и CMT2l (HSP22).
CMT3 или Dejerine-Sottas — тяжелая демиелинизирующая невропатия, которая начинается в младенчестве. У младенцев происходит тяжелая атрофия мышц, слабость и сенсорные проблемы. Это редкое расстройство может быть вызвано определенной точечной мутацией в гене P0 или точечной мутацией в гене PMP-22.
CMT4 включает несколько различных подтипов аутосомно-рецессивного демиелинизирующего двигателя и сенсорных невропатий. Каждый подтип невропатии вызван другой генетической мутацией, может влиять на конкретную этническую популяцию и производит различные физиологические или клинические характеристики. Лица с CMT4 обычно развивают симптомы слабости ног в детстве, а в подростковом возрасте они не могут ходить.
CMTX возникает точечной мутацией гена connexin-32 на Х-хромосоме. Коннексин-32-белок экспрессируется в клетках Schwann-клеток, которые обертывают нервные аксоны, составляя один сегмент миелиновой оболочки. Этот белок может участвовать в связи с клеткой Шванна с аксоном. Мужчины, которые наследуют один мутированный ген от своих матерей, проявляют умеренные или тяжелые симптомы заболевания, начиная с позднего детства или подросткового возраста (Y-хромосома, которую самцы наследуют от своих отцов, не имеет ген коннексин-32).
Процедуры и операции
Особое внимание уделяется немедикаментозным методам терапии. Комплекс мероприятий, позволяющих достичь максимального терапевтического эффекта:
- Лечебная физическая культура. Регулярные занятия ЛФК повышают мышечный тонус. Наибольший эффект достигается при достижении пассивных упражнений (занятия вместе со специалистом) и активных (выполняются самостоятельно).
- Электростимуляция. Направленная подача электрических импульсов улучшает проводимость по периферической нервной системе, активирует метаболические процессы в паретичных мышцах, усиливает нейротрофику.
- Бальнеотерапия. Грязевые аппликации и грязевые ванны замедляют формирование контрактур и улучшают работу вегетативной нервной системы.
- Массаж. Различные виды массажа (аппаратный и ручной) улучшают лимфатический отток и нормализуют кровообращение. Рекомендован расслабляющий, стимулирующий и вибрационный массаж.
- Ортопедическая терапия. Ношение специальной ортопедической обуви позволяет предотвратить развитие грубой деформации. Из-за мышечной слабости развивается нестабильность суставов. Подтяжки, ортезы и другие специальные приспособления используются для фиксации стоп в необходимом положении.
ЛЕКЦИИ ПО МЕДИЦИНСКОЙ ГЕНЕТИКЕ — PDF … ЛЕКЦИИ ПО МЕДИЦИНСКОЙ ГЕНЕТИКЕ — PDF … Рассекающий остеохондрит (РО): атлас …
При комплексном подходе к терапии повышается уровень мышечной силы, нормализуется походка и исправляются различные нарушения равновесия. Грамотно подобранное лечение повышает социальную и бытовую адаптацию, восстанавливает работоспособность пациента.
Мальформация Киари 2
Мальформация Киари 2 больше известна, как мальформация Арнольд-Киари, и является довольно частой врожденной патологией спинного мозга и задней черепной ямкой, при которой встречается миеломенингоцеле и каудальная дислокация в большое затылочное отверстие таких анатомических структур, как червя мозжечка, миндалин, четвертого желудочка, ствола мозга, продолговатого мозга.
Эпидемиология
Аномалия Киари 2 встречается 1:1000 новорожденным. Дети рожденные с миеломенингоцеле в 95% случаях имеют такую ассоциированную патологию, как мальформацию Киари 2.
Клинические проявления
Клиника зависит от возраста:
Неонатальный период
- миеломенингоцеле
- дисфункция ствола мозга
- нейрогенный мочевой пузырь
Детство
- гидроцефалия
- нарушение двигательной функции (вплоть до тетраплегии)
Взрослые
- Гидромиелия, сирингомиелия и сирингобульбия.
- Сколиоз
Патология
Данная патология обусловлена дислокацией в большое затылочное отверстие таких анатомических структур, как червя мозжечка, миндалин, четвертого желудочка, ствола мозга, продолговатого мозга в следствие недоразвития задней черепной ямки.
Ассоциация
Патология спинного мозга
- сирингомиелия
- сколиоз
- сегментарные аномалии до 50% случаев
- диастематомиелия (дипломиелия, раздвоение спинного мозга, удвоение спинного мозга).
Патология головного мозга
- дисгенезия мозолистого тела
- отсутствие прозрачной перегородки
- обструктивная гидроцефалия
- полимикрогирия при шизэнцефалии
- фенестрация серпа
Патология костей черепа
- задняя черепная ямка маленьких размеров
- расширение большого отверстия
- фестончатость пирамиды височной кости
Патология скелета
Радиологические находки
Антенатальное ультразвуковое исследование
- Фетальная вентрикуломегалия
- Дисгенезия мозолистого тела
- Симптом лимона— это признак, характеризующий изменение формы головы в виде банана.
- Симптом банана— это признак, который характеризуется патологическим изменённой формой мозжечка с уплощением задней поверхности вследствие облитерации большой цистерны.
МРТ
Задняя черепная ямка
- дислокация в большое затылочное отверстие таких анатомических структур, как червя мозжечка, миндалин, четвертого желудочка, ствола мозга, продолговатого мозга.
- обструктивная гидроцефалия. Стеноз на уровне водопровода.
- низкое положение намета мозжечка.
Спинной мозг
- симптом натянутого спинного мозга
- спина бифида — миеломенингоцеле
Дифференциальный диагноз
- Мальформация Киари 1 (нет миеломенингоцеле).
- Изолированное миеломенингоцеле без аномалии задней черепной ямки.
Источник
3. Naidich TP, Pudlowski RM, Naidich JB et-al. Computed tomographic signs of the Chiari II malformation. Part I: Skull and dural partitions. Radiology. 1980;134 (1): 65-71. Radiology (abstract) — Pubmed citation
4. Curnes JT, Oakes WJ, Boyko OB. MR imaging of hindbrain deformity in Chiari II patients with and without symptoms of brainstem compression. AJNR Am J Neuroradiol. 10 (2): 293-302. AJNR Am J Neuroradiol (abstract) — Pubmed citation
5. Naidich TP, Mclone DG, Fulling KH. The Chiari II malformation: Part IV. The hindbrain deformity. Neuroradiology. 1983;25 (4): 179-97. — Pubmed citation
6. Dipietro MA, Venes JL, Rubin JM. Arnold-Chiari II malformation: intraoperative real-time US. Radiology. 1987;164 (3): 799-804. Radiology (abstract) — Pubmed citation
7. Maixner WJ. Spina Bifida, Management and Outcome. Springer. (2008) ISBN:8847006511. Read it at Google Books — Find it at Amazon
8. Osborn A, Blaser S, Salzman K. Encyclopedia of Diagnostic Imaging. AMIRSYS. (2008) ISBN:0721629059. Read it at Google Books — Find it at Amazon
Читайте также: