Микроделеционные и микродупликационные синдромы
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Микроделеционные и микродупликационные синдромы (ММДС) представляют собой клинически гетерогенную группу заболеваний, которые, однако, характеризуются потерей или приобретением части хромосомы. Данную аберрацию невозможно детектировать с помощью классического кариотипирования, и единственным лабораторным методом его подтверждения является молекулярное генотипирование. На данный момент описано более 30 специфических рекуррентных ММДС.
В основе патогенеза данных болезней лежит комплексное нарушение функции белков, гены которых располагаются в поврежденном участке. Заболевания группы ММДС характеризуются очень разнообразной симптоматикой, однако имеют много общих черт: отставание интеллектуального и физического развития, множественные пороки развития, лицевые дисморфизмы, судорожные припадки. Несмотря на то, что каждый синдром имеет ряд характерных симптомов, иногда очень сложно подтвердить диагноз, основываясь только на клинических проявлениях. Использование комплексного молекулярно-генетического скрининга на наиболее распространенные 30 синдромов позволяет значительно упросить первичное обследование пациентов с отставанием развития и пороками развития.
Проявления ММДС могут очень сильно варьировать у разных пациентов. Молекулярный скрининг на микроделеции/микродупликации хромосом выявляет 30 наиболее распространенных ММДС.
Необходимо отметить, что применяемый в данном случае метод диагностики позволяет выявить только несбалансированные микроделеции/дупликации. Поэтому использовать данный тест целесообразно только при наличии клинических признаков заболевания.
Носительство сбалансированных транслокаций, включающих указанные выше микроделеции/дупликации у клинически здоровых людей данный метод диагностики не покажет.
2q32-q33 микроделеционный синдром (SATB2 – ассоциированный синдром), 3q29 микроделеционный/микродупликационный синдром,
8q24.11-24.13 микроделеционный синдром (синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа),
17p13.3 микроделеционный синдром (LIS1-ассоциированная лиссэнцефалия, синдром Миллера-Дикера/ изолированная лиссэнцефалия/ синдром двойной коры),
22q11.2 микроделеционный синдром ( синдром ДиДжорджи, велокардиофациальный синдром ) синдром дупликации 15q,
Синдром 1р36 микроделеции клинически проявляется отставанием в развитии, мышечным гипотонусом, черепно-лицевыми аномалиями, брахи- и камптодактилией и укороченными нижними конечностями, а также возможны судорожные припадки, структурные аномалии мозга, врожденные пороки сердца, зрительные и глазные нарушения, тугоухость, аномалии развития скелета, наружных половых органов и почек.
Синдром 2q23.1 микроделеций/микродупликаций характеризуется общим отставанием в развитии, тяжелыми нарушениями речи, припадками, нарушениями сна, аутистическим поведением, намеренным нанесением себе телесных повреждений и агрессивным поведением. К другим клиническим признакам относятся микроцефалия, широкий рот, вздернутая верхняя губа, выдающиеся резцы, опущенные уголки рта, макроглоссия, аномалии развития уха.
Синдром 3q29 микроделеции/микродупликации: отставание в развитии, микроцефалия и офтальмологические нарушения, аномалии развития сердца, мышечный гипотонус, задержка речевого развития, краниосиностоз, высокое «готическое» небо, зубочелюстные аномалии, кондуктивная тугоухость, аномалии опорно-двигательного аппарата; припадки. Часто у многих носителей данной дупликации не наблюдается выраженной симптоматики, что связано со сниженной пенетрантностью.
Синдром Вольфа-Хиршхорна характеризуется типичными черепно-лицевыми аномалиями, пренатальным дефицитом роста, за которым следуют задержка постнатального развития и гипотонус мышц в сочетании с их недоразвитием. Также наблюдается отставание в общем развитии различной степени выраженности, судорожные припадки. К другим симптомам относят аномалии развития скелета, врожденные пороки сердца, глухота (в большинстве случаев кондуктивная, аномалии развития урогенитального тракта, структурные аномалии мозга).
Синдром кошачьего крика: высокочастотный монотонный плач, микроцефалия, широкая переносица, эпикантус, микрогнатия, измененная дерматоглифика, а также тяжелые психомотрные нарушения и умственная отсталость. Редко встречаются аномалии развития сердца, почек, возможно наличие преаурикулярных выростов, синдактилии, гипоспадии и крипторхизма. Клиническая симптоматика зависит от размера делеции и может сильно варьировать.
Синдром Сотоса характеризуется тремя важнейшими клиническими проявлениями: специфический внешний вид, избыточный рост, трудности в обучении. К другим симптомам относятся поведенческие нарушения, раннее окостенение, пороки сердца, аномалии черепа и почек, повышенная гибкость суставов, плоскостопие, сколиоз, неонатальная желтуха, гипотонус мышц, припадки.
Синдром Вильямса-Бойрена (7q11.23 дупликационный синдром) характеризуется повреждениями со стороны сердечно-сосудистой системы, соединительнотканной дисплазией, неврологическими нарушениями, нарушениями речи, поведенческими нарушениями, умственной отсталостью, эндокринными нарушениями.
Синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа) характеризуется особенностями развития эктодермы (мелкие редкие депигментированные и медленно растущие волосы, ониходистрофия, микромастия), а также деформацией скелета, множественными остеохондромами и высоким риском умственной отсталости легкой и средней степени выраженности.
Синдром 9q22.3 микроделеции приводит к развитию синдрома Горлина (синдром невоидной базальноклеточной карциномы), также возможны отставание в развитии, метопический краниосиностоз, обструктивная гидроцефалия, пре- и постнатальная макросомия, припадки. Симптомы 9q22.3 микроделеционного синдрома крайне вариабельны и зависят от размера микроделеции, который может достигать 270 генов.
Синдром ДиДжорджи/Велокардиофациальный синдром клинически характеризуется отставанием развития, болезни аутистического спектра врожденными пороками сердца, дефектами неба и характерными чертами. Помимо этого имеется аплазия тимуса, ведущая к иммунодефициту, и паращитовидных желез, ведущая к гипокальциемии, а также нарушения кормления и глотания и множественные пороки развития, судорожные припадки.
Синдром дупликации 15q проявляется отставаниями в языковом развитии и моторных навыках, таких как ходьба и сидение, гипотонией, припадками, низкорослостью. Отличительными признаками являются очень тонкие черты лица, однако могу присутствовать такие признаки, как эпикантальные складки, широкий лоб, сплющенный мост носа, нос «кнопкой», высокое арочное небо. У многих пациентов наблюдаются проявления заболеваний аутистического спектра, такие как нарушения коммуникации и социальных взаимодействий, навязчивые интересы, нарушенные циклы сна и повторяющиеся и стереотипное поведение.
Синдром Прадера-Вилли характеризуется мышечным гипотонусом, нарушением кормления в период младенчества, склонностью к перееданию в период раннего детства и постепенным развитием морбидного ожирения. Имеется отставание нормальных этапов речевого и моторного развития. В той или иной степени у всех пациентов имеются когнитивные расстройства. Специфичен поведенческий фенотип, проявляющийся в виде истерик (tempertantrum), упрямства, манипулятивного поведения, обсессивно-компульсивных растройств. Для пациентов обоих полов характерен гипогонадизм, проявляющийся в виде гипоплазии половых органов, неполноценности полового созревания, а также бесплодие. При отсутствии лечения соматотропным гормоном характерен невысокий рост. К другим внешним проявлениям относятся страбизм, сколиоз.
Синдром Ангельмана характеризуется тяжелым отставанием в развитии и умственной неполноценностью, нарушением речи, атактической походкой и/или тремором конечностей, а также уникальным поведенческим паттерном (частый смех, улыбка, возбудимость), который выявляется не раньше первого года жизни. Отставание в развитии обычно обнаруживается в первые полгода жизни. Зачастую правильный диагноз удается поставить только через несколько лет.
Микроделеционные и микродупликационные синдромы
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Синонимы русские
Микроделеции и микродупликации.
Синонимы английские
Microdeletion and microduplication.
Метод исследования
Полимеразная цепная реакция, фрагментный анализ, MLPA.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Синдромы данной группы чаще всего характеризуются нарушением когнитивных функций, задержкой речевого развития и задержкой роста, различного рода стигмами, дисморфизмами и мальформациями, затрагивающими широкий спектр систем и органов. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Исследование на основные формы микроделеционных и микродупликационных синдромов позволяет комплексно и одновременно выявлять 26 синдромов, такие как синдром ДиДжорджи, синдром Прадера - Вилли, синдром Ангельмана, синдром кошачьего крика, 1р36 делеция и другие. Рекомендуется проводить данное исследование в качестве первичного скрининга у пациентов с синдромальными и несиндромальными формами отставания в развитии.
Данный тест детектирует мутации, характерные для следующих нозологий: 1р36 микроделеционный синдром, 2p16.1-p15 микроделеционный синдром, 2q23.1 микроделеционный/микродупликационный синдром, 3q29 микроделеционный/микродупликационный синдром, 9q22.3 микроделеционный синдром, LIS1-ассоциированная лиссэнцефалия (синдром Миллера - Дикера / изолированная лиссэнцефалия / синдром двойной коры), SATB2 - ассоциированный синдром, нейрофиброматоз 1-го типа, KANSL1-связанная умственная отсталость, синдром Виттевеена - Колька, синдром Вольфа - Хиршхорна, синдром ДиДжорджи / велокардиофациальный синдром, синдром дупликации 15q, синдром дупликации гена MECP2, синдром кошачьего крика, синдром Лангера - Гидеона (трихоринофалангеальный синдром 2-го типа), синдром Прадера - Вилли / синдром Ангельмана, синдром Рубинштейна - Тейби, синдром Смита - Магениса, синдром Сотоса, синдром Фелана - МакДермида, синдром Вильямса - Бойрена, синдром Потоцки - Лупски, синдром Клайнфельтера, синдром Шерешевского - Тернера, синдром тройной Х хромосомы.
Для чего используется исследование?
- Диагностика микроделеционных и микродупликационных заболеваний
Когда назначается исследование?
- При подтверждении причин отставания и задержки развития.
- При дифференциальной диагностике причин пороков развития сердца, печени, почек, нервной системы.
- При подтверждении диагноза "микроделеционный" или "микродупликационный синдром".
Что означают результаты?
Наличие патогенной микроделеции или микродупликации подтверждает диагноз "ММС".
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды и индивидуальных генетических факторов.
Научная электронная библиотека

Ранее, при исследовании кариотипа классическими цитогенетическими методами, считалось, что структурные аномалии хромосом (инсерции, инверсии, делеции, дупликации, транслокации) встречаются значительно реже, чем численные. Выявление структурных аномалий хромосом зависит от их размера, локализации, типа перестройки (регулярная или мозаичная), а также метода, используемого для исследования. При этом разрешающая способность классических цитогенетических методов составляет от 5–7 млн пн. С внедрением современных молекулярно-цитогенетических технологий исследования хромосом (генома) стали возможны с разрешающей способностью от 1000 пн и меньше, что позволяет обнаружить субмикроскопические перестройки и уточнить координаты геномных нарушений, в том числе микроделеций и микродупликаций, и число обнаруженных структурных перестроек, значительно возросло. Это, в свою очередь, позволяет получать информацию о генах, находящихся в исследуемых участках, и в дальнейшем проводить биоинформатический анализ с целью определения генов-кандидатов патологических фенотипических проявлений у детей.
Данные аномалии часто выявляются при анализе генома детей с недифференцированными формами умственной отсталости, микроаномалиями развития (МАР) и врождёнными пороками развития (ВПР). Известно, что структурные аномалии хромосом ассоциированы с определёнными фенотипическими проявлениями, в том числе, как и с наиболее частыми микроделеционными/микродупликационными синдромами, так и с редкими микроаномалиями. Несмотря на это, даже при наиболее часто встречающихся микроделеционных/микродупликационных синдромах, затрагивающих области, в которых локализованы более 100 генов, вклад отдельных генов в формирование патологических фенотипических проявлений ещё предстоит изучать. Наиболее часто встречающиеся микроделеционные/микродупликационные синдромы и аномалии представлены в таблице 2. У многих синдромов, описанных с внедрением современных молекулярно-цитогенетических технологий, частота не известна. Ниже даётся описание отдельных микроделеционных/микродупликационных синдромов и аномалий.
Синдром микроделеции 1p36
При данном синдроме наблюдаются задержка моторного развития, расстройства аутистического спектра; некоторые исследователи отмечают, что 25 % пациентов могут ходить самостоятельно, широкой походкой, примерно к 2–7 годам. Экспрессивная речь отсутствует в 75 % случаев, понимание обращенной речи ограничено определенными ситуациями. Стремление к коммуникации в ранние периоды развития проявляется слабо, но улучшается со временем при расширении используемых жестов. Многие исследователи выявляют следующие клинические признаки: резкие перемены настроения, самоповреждающее поведение, стереотипии и МАР: прямые брови, микроцефалия, широкая переносица, низко расположенные аномальной формы ушные раковины, клинодактилия, небольшие ступни, а также отмечают пороки сердечно-сосудистой системы (ССС).
При микроделеции 1q41q42 наблюдаются умеренная умственная отсталость, аутизм, судороги, микроцефалия, косолапость, МАР: гипотелоризм глазных щелей, вывернутые вперёд ноздри.
Синдром микроделеции 2q37
Описано более 100 клинических случаев с микроделецией участка 2q37. У этих индивидуумов обычно отмечается лёгкая или умеренная задержка психического развития, у 30 % пациентов имеются черты аутизма. Дети обычно невысокого роста с гипермобильными суставами, сколиозом, синдактилией кистей и/или стоп. Пороки сердца встречаются у 35 %, нередко выявляются пороки почек. К характерным лицевым аномалиям при синдроме микроделеция 2q37 относят следующие: брахицефалия, круглое широкое лицо, редкие волосы на голове, широкие лоб и нос с расщеплением кончика, глубоко посаженные глаза, редкие высокие арочные брови, готическое нёбо; отмечают также укороченные фаланги пальцев рук и ног, экзему.
При данной микроделеции наблюдается умственная отсталость средней тяжести, отмечаются аутистические черты, МАР: микроцефалия, длинное узкое лицо, большие ушные раковины, аномально сформированная спинка носа, а также атаксия, пороки сердца и почек. Нередко отмечают аутистические проявления.
При данной микродупликации наблюдается умственная отсталость средней тяжести, встречаются аутистические проявления, задержка роста, задержки психомоторного (ЗПМР) и психоречевого развития (ЗПРР), микроцефалия, лицевые аномалии: высокий лоб, низко расположенные ушные раковины, короткая шея, аномалии глазных щелей, а также гипотония с мышечной гипотрофией, пороки ССС.
Синдром Вольфа-Хиршхорна (делеция в участке 4р16)
При данной микроделеции наблюдаются умственная отсталость различной степени тяжести, задержка роста, ЗПМР и ЗПРР, микроцефалия, лицевые микроаномалии: высокий лоб, эпикант, маленький рот с опущенными углами, клювовидный нос с выступающим надпереносьем, микрогения, низко расположенные деформированные ушные раковины с преарикулярными складками, короткая шея, а также пороки ССС, почек, желудочно-кишечного тракта, гипоспадия, гипотония с мышечной гипотрофией, судороги. Иногда встречаются пороки мозга в виде агенезии или гипоплазии мозолистого тела, гипоплазии мозжечка. Характерным признаком синдрома является воронкообразное углубление в области крестца (sinus sacralis). Частота этого синдрома в популяции 1:90000 – 100000 по данным публикаций различных авторов. Встречаются аутистические расстройства.
Синдром «крика кошки» («cri du chat», делеция в участке 5p15.2)
Синдромы Вольфа-Хиршхорна и «крика кошки» относятся к делеционным синдромам, но довольно часто геномные микроаномалии затрагивают только критические сегменты хромосом (4р16 и 5р15, соответственно), и тогда можно их отнести к микроделеционным синдромам.
При данной микродупликации наблюдаются умственная отсталость различной степени тяжести, аутизм, черепно-лицевые аномалии: макроцефалия, гипертелоризм глазных щелей, аномалии ушных раковин.
Синдром Вильямса (делеция в участке 7q11.23)
Основными проявлениями синдрома являются хрипловатый голос, отсутствие чувства дистанции при общении. У пациентов с синдромом Вильямса наблюдается слабая зрительно-моторная интеграция, в результате чего вместо целостной картинки они видят ее отдельные составные части. Кроме того, у больных выявлены макроцефалия, макрокрания, мышечная гипотония, МАР: широкий лоб, глубоко посаженные глазные щели, широкая короткая переносица, оттопыренные ушные раковины, колобома, аномалии зубов; потеря слуха, тревожность, дефицит внимания с гиперактивностью (СДВГ), пороки ССС. Многие дети с этим синдромом могут играть на музыкальных инструментах, общительны, не имеют задержки в речевом развитии. Частота 1:10000 – 20000 по данным публикаций различных авторов.
При данной микроделеции наблюдаются ЗПРР, расстройства аутистического спектра, МАР: синофриз, широкий нос, ретрогнатия, широкие пальцы с короткими дистальными фалангами.
Синдром микродупликации 15q11q13
Следует отметить, что микродупликацию 15q11q13 можно отнести к часто встречающимся цитогенетическим аномалиям при аутизме. По данным литературы, указанная микроперестройка встречается примерно у 1 % детей с аутизмом, и она индексирована в базе данных OMIM (Online Mendelian Inheritance in Man) (OMIM:608636) как генетически обусловленное состояние. Имеются исследования, направленные на приоритизацию генов-кандидатов психических нарушений у пациентов с перестройками в этом участке, включая и микродупликацию.
Синдром Ангельмана (делеция или унипарентальная дисомия в участке 15q11.2q13 материнского происхождения)
Нарушения в данном участке связаны с таким генетическим заболеванием, как синдром Ангельмана при нарушении в хромосоме материнского происхождения. При перестройке материнского происхождения у детей часто наблюдаются эпилепсия, атаксия, умственная отсталость и характерные лицевые микроаномалии. Основной значимой характеристикой синдрома Ангельмана является чрезмерно положительное настроение с постоянной улыбкой и смехом у ребёнка. Выявляются фокальные стереотипии, однако не наблюдаются специфичные, часто повторяющиеся стереотипные движения. Дети заинтересованы в социальных взаимодействиях, многие пациенты стремятся к коммуникации, несмотря на выраженные нарушения речи. Пациенты испытывают трудности во взаимодействии по причине слабого понимания социальных и эмоциональных сигналов. Психические нарушения зависят от происхождения перестройки, а именно, от того, является ли делетированный участок материнским или отцовским. Это позволяет говорить о том, что эпигенетический феномен геномного импринтинга играет значимую роль в этиологии и патогенезе психических нарушений.
Синдром Прадера-Вилли (делеция или унипарентальная дисомия в участке 15q11.2q13 отцовского происхождения)
Нарушения поведения при данном синдроме проявляются в резких перепадах настроения, упорстве, соответствующем поведении, обсессивно-компульсивных характеристиках, а также сложностью в отвлечении от ежедневно повторяемых рутинных событий. Синдром Прадера-Вилли ассоциирован с повышенным риском психических нарушений. Пациенты с синдром Прадера-Вилли при отсутствии отцовской копии указанного участка хромосомы 15 (унипарентальная дисомия) подвержены различным психическим расстройствам.
При данной микроделеции наблюдаются умственная отсталость, низкорослость, микроцефалия, пониженный мышечный тонус (гипотония), скелетные аномалии, гипоспадия.
Синдром микроделеции 16p11.2
Перестройки в участке 16p11.2, по данным литературы, ассоциированы с аутизмом и лёгкой формой умственной отсталости. При исследовании детей с расстройствами аутистического спектра микроделеция участка 16p11.2 выявляется в 0,4–1,2 % случаев. Чаще обнаруживают эту микроделецию de novo; тем не менее, микроделеция также может передаваться от родителей к ребёнку. Микроделеции участка 16p11.2 встречаются чаще при аутизме по сравнению с микродупликациями этого же участка. У пациентов с микроделецией в данном участке отмечают более тяжёлые фенотипические проявления по сравнению с микродупликацией. Выявляются следующие врождённые проявления: задержка речевого развития (ЗРР), отставание в физическом развитии, двигательные нарушения, эпилепсия, пороки сердца, ожирение. Среди МАР отмечают низко расположенные ушные раковины и частично перепончатые пальцы.
Синдром Смит-Маженис (делеция в участке 17p11.2)
Для микроделеции участка 17p11.2 характерны различные фенотипические проявления. Эти признаки были объединены в синдром Смит-Маженис, индексированным в OMIM (182290). Для синдрома характерны черепно-лицевые аномалии (широкое/плоское лицо, широкая переносица, выпуклый лоб, сросшиеся брови, низко расположенные ушные раковины), широкие короткие кисти, плоскостопие, брахидактилия, а также нарушения развития и поведения. Черты аутизма присутствуют более чем у 50 % детей с данным синдромом. У 80 % пациентов проявляется самоповреждающее поведение (аутоагрессия), включая онихотилломанию, кусание запястий, качание головой, а также повышенную толерантность к боли, нарушение сна. Навыки импрессивной речи, как правило, выше, чем экспрессивной. Хриплый голос может являться диагностическим маркером синдрома. Использование языка жестов значительно способствует улучшению коммуникативных способностей ребенка. Выявляют умственную отсталость от умеренной до тяжёлой степени.
Следует сказать и про микродупликацию в этом участке, которая ассоциирована с синдромом Потоцки-Лупски, индексированным в OMIM (610883). Заболевание проявляется гипотонией мышц, врождёнными пороками развития, лицевыми аномалиями, пороками ССС, низким ростом и различными нервными и психическими нарушениями. По данным исследований, аутизм встречается у этих детей в 65 % случаев.
Синдром микроделеции 22q11.2
Синдром микроделеции участка 22q11.2 ассоциируется с аутизмом, велокардиофациальным синдромом и синдромом ДиДжорджи. Частота его составляет 1:4000 новорождённых. У детей с такой перестройкой выявляют нарушение психического и моторного развития, отмечают медленный рост и гипотонию. К менее распространённым нарушениям при нём можно отнести аномалии ССС, расщелину нёба, лицевые МАР, нарушение иммунитета. Аутистические проявления наблюдаются также у 15–25 % пациентов с микродупликацией в данном участке.
Синдром микроделеции 22q13.3
Синдром микроделеции 22q13.3 известен также как синдром Фелан-МакДермид. Он с одинаковой частотой встречается у лиц мужского и женского пола. К характерным признакам данного синдрома можно отнести неонатальную гипотонию, микроаномалии развития, задержку или полное отсутствие речи, аутизм и эпилепсию. К основным фенотипическим проявлениям данного синдрома относятся следующие: большие ушные раковины, диспластичные ногти, широкий лоб, острый подбородок. При исследовании нескольких групп детей с микроделецией в участке 22q13.3 аутистические черты были выявлены у 85 % пациентов.
Основные характерные особенности, свойственные известным микроделеционным и микродупликационным синдромам, представлены в таблице 2.
Характерные особенности некоторых часто встречающихся микроделеционных* и микродупликационных* синдромов и аномалий
Санкт-Петербургское государственное
бюджетное учреждение здравоохранения
«Родильный дом №17»
Среди клинически значимых хромосомных болезней у человека синдромы микроделеций имеют немаловажное значение.
По данным ВОЗ эта группа хромосомных аномалий вносит значимый вклад в структуру умственной отсталости. Микроделеционные синдромы (МДС) - это особый вид хромосомных заболеваний, при котором происходит потеря микроскопического участка хромосомного материала, что не удается зафиксировать рутинными методами цитогенетической диагностики.
К наиболее часто встречающимся МДС относятся CATCH-синдром, синдром микроделеции 1p36, синдром Прадера-Вилли, синдром Ангельмана, синдром Вольфа-Хиршхорна и другие. Актуальность МДС связана с рядом причин.
Во-первых, МДС являются тяжелыми заболеваниями, которые сопровождаются задержкой физического и психического развития (рис. 1.)
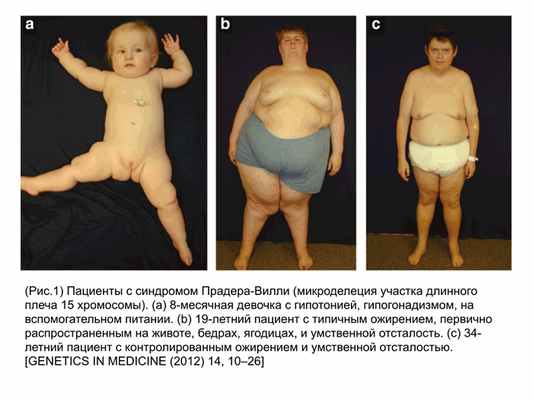
Во-вторых, МДС встречаются с достаточно высокой частотой. Так, например, средняя частота встречаемости 5 наиболее распространенных МДС составляет 1:1000 новорожденных, что сопоставимо со средней популяционной частотой встречаемости синдрома Дауна. Некоторые МДС встречаются в популяции гораздо чаще, чем многие хромосомные и генные заболевания.
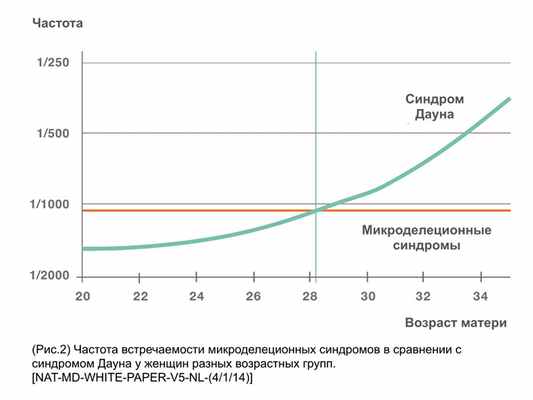
В третьих, в отличие от таких хромосомных заболеваний как синдром Дауна, синдром Эдвадса, синдром Патау, синдром Тернера, при которых важным фактором риска рождения больного ребенка является возраст матери, риск появления МДС не зависит от возраста женщины. Таким образом, женщины до 30 лет имеют более высокую вероятность рождения детей с микроделеционными синдромами, чем вероятность рождения детей с синдромом Дауна (рис. 2.)
В четвертых, очень большое количество случаев МДС остается не диагностированным во время беременности и после рождения ребенка.
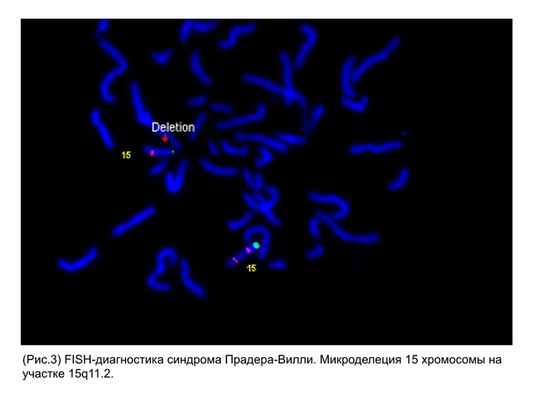
Тем не менее, многие из известных МДС могут проявляться во время беременности ультразвуковыми маркерами хромосомных болезней, и поэтому могут быть заподозрены уже антенатально в ходе проведения ультразвукового исследования. Однако, стандартом диагностики МДС является инвазивная пренатальная диагностика с применением специальных методов цитогенетического анализа (FISH-диагностика) (рис. 3).
На базе центра пренатальной диагностики Родильного дома N17 возможно обследование беременных женщин с целью исключения МДС у плода:
- В случае, если у женщины имеются показания к проведению инвазивного исследования, возможно выполнение расширенного цитогенетиечского анализа, включающего стандартную цитогенетическую диагностику и диагностику на МДС.
- В случае, если у женщины нет показаний для инвазивной пренатальной диагностики возможно исследовать МДС с помощью неинвазивных методов пренатальной диагностики, при которых анализируется ДНК плода в крови матери.
Молекулярный скрининг на микроделеции/микродупликации хромосом (Microdeletion And Microduplication Syndromes)
Микроделеционные и микродупликационные синдромы (ММДС) представляют собой клинически гетерогенную группу заболеваний, которые, однако, характеризуются потерей или приобретением части хромосомы. Данную аберрацию невозможно детектировать с помощью классического кариотипирования, и единственным лабораторным методом его подтверждения является молекулярное генотипирование. На данный момент описано более 30 специфических рекуррентных ММДС.
В основе патогенеза данных болезней лежит комплексное нарушение функции белков, гены которых располагаются в поврежденном участке. Заболевания группы ММДС характеризуются очень разнообразной симптоматикой, однако имеют много общих черт: отставание интеллектуального и физического развития, множественные пороки развития, лицевые дисморфизмы, судорожные припадки. Несмотря на то, что каждый синдром имеет ряд характерных симптомов, иногда очень сложно подтвердить диагноз, основываясь только на клинических проявлениях. Использование комплексного молекулярно-генетического скрининга на наиболее распространенные 30 синдромов позволяет значительно упросить первичное обследование пациентов с отставанием развития и пороками развития.
Проявления ММДС могут очень сильно варьировать у разных пациентов. Молекулярный скрининг на микроделеции/микродупликации хромосом выявляет 30 наиболее распространенных ММДС.
2q32-q33 микроделеционный синдром (SATB2 – ассоциированный синдром), 3q29 микроделеционный/микродупликационный синдром,
8q24.11-24.13 микроделеционный синдром (синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа),
17p13.3 микроделеционный синдром (LIS1-ассоциированная лиссэнцефалия, синдром Миллера-Дикера/ изолированная лиссэнцефалия/ синдром двойной коры),
22q11.2 микроделеционный синдром ( синдром ДиДжорджи, велокардиофациальный синдром ) синдром дупликации 15q,
Синдром 1р36 микроделеции клинически проявляется отставанием в развитии, мышечным гипотонусом, черепно-лицевыми аномалиями, брахи- и камптодактилией и укороченными нижними конечностями, а также возможны судорожные припадки, структурные аномалии мозга, врожденные пороки сердца, зрительные и глазные нарушения, тугоухость, аномалии развития скелета, наружных половых органов и почек.
Синдром 2q23.1 микроделеций/микродупликаций характеризуется общим отставанием в развитии, тяжелыми нарушениями речи, припадками, нарушениями сна, аутистическим поведением, намеренным нанесением себе телесных повреждений и агрессивным поведением. К другим клиническим признакам относятся микроцефалия, широкий рот, вздернутая верхняя губа, выдающиеся резцы, опущенные уголки рта, макроглоссия, аномалии развития уха.
Синдром 3q29 микроделеции/микродупликации: отставание в развитии, микроцефалия и офтальмологические нарушения, аномалии развития сердца, мышечный гипотонус, задержка речевого развития, краниосиностоз, высокое «готическое» небо, зубочелюстные аномалии, кондуктивная тугоухость, аномалии опорно-двигательного аппарата; припадки. Часто у многих носителей данной дупликации не наблюдается выраженной симптоматики, что связано со сниженной пенетрантностью.
Синдром Вольфа-Хиршхорна характеризуется типичными черепно-лицевыми аномалиями, пренатальным дефицитом роста, за которым следуют задержка постнатального развития и гипотонус мышц в сочетании с их недоразвитием. Также наблюдается отставание в общем развитии различной степени выраженности, судорожные припадки. К другим симптомам относят аномалии развития скелета, врожденные пороки сердца, глухота (в большинстве случаев кондуктивная, аномалии развития урогенитального тракта, структурные аномалии мозга).
Синдром кошачьего крика: высокочастотный монотонный плач, микроцефалия, широкая переносица, эпикантус, микрогнатия, измененная дерматоглифика, а также тяжелые психомотрные нарушения и умственная отсталость. Редко встречаются аномалии развития сердца, почек, возможно наличие преаурикулярных выростов, синдактилии, гипоспадии и крипторхизма. Клиническая симптоматика зависит от размера делеции и может сильно варьировать.
Синдром Сотоса характеризуется тремя важнейшими клиническими проявлениями: специфический внешний вид, избыточный рост, трудности в обучении. К другим симптомам относятся поведенческие нарушения, раннее окостенение, пороки сердца, аномалии черепа и почек, повышенная гибкость суставов, плоскостопие, сколиоз, неонатальная желтуха, гипотонус мышц, припадки.
Синдром Вильямса-Бойрена (7q11.23 дупликационный синдром) характеризуется повреждениями со стороны сердечно-сосудистой системы, соединительнотканной дисплазией, неврологическими нарушениями, нарушениями речи, поведенческими нарушениями, умственной отсталостью, эндокринными нарушениями.
Синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа) характеризуется особенностями развития эктодермы (мелкие редкие депигментированные и медленно растущие волосы, ониходистрофия, микромастия), а также деформацией скелета, множественными остеохондромами и высоким риском умственной отсталости легкой и средней степени выраженности.
Синдром 9q22.3 микроделеции приводит к развитию синдрома Горлина (синдром невоидной базальноклеточной карциномы), также возможны отставание в развитии, метопический краниосиностоз, обструктивная гидроцефалия, пре- и постнатальная макросомия, припадки. Симптомы 9q22.3 микроделеционного синдрома крайне вариабельны и зависят от размера микроделеции, который может достигать 270 генов.
Синдром ДиДжорджи/Велокардиофациальный синдром клинически характеризуется отставанием развития, болезни аутистического спектра врожденными пороками сердца, дефектами неба и характерными чертами. Помимо этого имеется аплазия тимуса, ведущая к иммунодефициту, и паращитовидных желез, ведущая к гипокальциемии, а также нарушения кормления и глотания и множественные пороки развития, судорожные припадки.
Синдром дупликации 15q проявляется отставаниями в языковом развитии и моторных навыках, таких как ходьба и сидение, гипотонией, припадками, низкорослостью. Отличительными признаками являются очень тонкие черты лица, однако могу присутствовать такие признаки, как эпикантальные складки, широкий лоб, сплющенный мост носа, нос «кнопкой», высокое арочное небо. У многих пациентов наблюдаются проявления заболеваний аутистического спектра, такие как нарушения коммуникации и социальных взаимодействий, навязчивые интересы, нарушенные циклы сна и повторяющиеся и стереотипное поведение.
Синдром Прадера-Вилли характеризуется мышечным гипотонусом, нарушением кормления в период младенчества, склонностью к перееданию в период раннего детства и постепенным развитием морбидного ожирения. Имеется отставание нормальных этапов речевого и моторного развития. В той или иной степени у всех пациентов имеются когнитивные расстройства. Специфичен поведенческий фенотип, проявляющийся в виде истерик (tempertantrum), упрямства, манипулятивного поведения, обсессивно-компульсивных растройств. Для пациентов обоих полов характерен гипогонадизм, проявляющийся в виде гипоплазии половых органов, неполноценности полового созревания, а также бесплодие. При отсутствии лечения соматотропным гормоном характерен невысокий рост. К другим внешним проявлениям относятся страбизм, сколиоз.
Синдром Ангельмана характеризуется тяжелым отставанием в развитии и умственной неполноценностью, нарушением речи, атактической походкой и/или тремором конечностей, а также уникальным поведенческим паттерном (частый смех, улыбка, возбудимость), который выявляется не раньше первого года жизни. Отставание в развитии обычно обнаруживается в первые полгода жизни. Зачастую правильный диагноз удается поставить только через несколько лет.
Литература
- Hague J, Wynn S, Middlemiss P The emerging microdeletion and microduplication syndromes: the importance of a diagnosis for both professionals and families Archives of Disease in Childhood 2012;97:A160-A161.
- Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena Jr., David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova, and Thomas Liehr. Microdeletion and Microduplication Syndromes. Journal of Histochemistry & Cytochemistry 60(5) 346–358, 2012.
Специальной подготовки к исследованию не требуется. Взятие крови желательно проводить не ранее 4 часов после приема пищи.
Литература
- Hague J, Wynn S, Middlemiss P The emerging microdeletion and microduplication syndromes: the importance of a diagnosis for both professionals and families Archives of Disease in Childhood 2012;97:A160-A161.
- Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena Jr., David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova, and Thomas Liehr. Microdeletion and Microduplication Syndromes. Journal of Histochemistry & Cytochemistry 60(5) 346–358, 2012.
- Заболевания аутистического спектра
- Множественные пороки развития: сердца, почек, легких, нервно-мышечной системы
- Иммунодефицит
- При подозрении на специфический микродупликационный/микроделеционный синдром.
Литература
- Hague J, Wynn S, Middlemiss P The emerging microdeletion and microduplication syndromes: the importance of a diagnosis for both professionals and families Archives of Disease in Childhood 2012;97:A160-A161.
- Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena Jr., David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova, and Thomas Liehr. Microdeletion and Microduplication Syndromes. Journal of Histochemistry & Cytochemistry 60(5) 346–358, 2012.
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Читайте также: