Мышечные дистрофии. Мышечная дистрофия Дюшенна. Мышечная дистрофия Беккера.
Добавил пользователь Валентин П. Обновлено: 27.01.2026
(Мышечная дистрофия Дюшенна; мышечная дистрофия Беккера)
, MDCM, New York Presbyterian Hospital-Cornell Medical Center
Мышечные дистрофии — это группа наследственных заболеваний мышц, при которых один или несколько генов, необходимых для нормальной мышечной структуры и функции, являются дефектными, что приводит к мышечной слабости Слабость Слабость — это потеря мышечной силы. То есть люди не могут нормально двигать мышцами, сколько бы не старались. Тем не менее, этот термин зачастую употребляется неправильно. Многие люди с нормальной. Прочитайте дополнительные сведения различной степени тяжести. Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера вызывают слабость в мышцах, расположенных ближе к туловищу.
Эти дистрофии вызваны дефектами в генах, отвечающих за функцию мышц, которые приводят к мышечной слабости, развивающейся в детском или подростковом возрасте и почти всегда возникающей у мальчиков.
Обе дистрофии характеризуются физической слабостью.
Диагноз основывается на результатах анализов крови и образца мышечной ткани.
Лечение включает физиотерапию и, в некоторых случаях, хирургическое вмешательство при обоих типах дистрофии, а также преднизон или дефлазакорт при дистрофии Дюшенна.
Мышечная дистрофия Дюшенна является второй по частоте (наиболее распространенной является плече-лопаточно-лицевая дистрофия Плече-лопаточно-лицевая мышечная дистрофия Плече-лопаточно-лицевая мышечная дистрофия является наиболее распространенной формой мышечной дистрофии. Поражаются мышцы лица и плеча. Мышечные дистрофии — это группа наследственных заболеваний. Прочитайте дополнительные сведения ) и самой тяжелой формой мышечной дистрофии. Болезнь начинается в раннем детстве. Мышечная дистрофия Беккера, хотя и тесно связана с мышечной дистрофией Дюшенна, начинается позже, в подростковом возрасте, и вызывает более легкие симптомы. Эти дистрофии почти всегда возникают у мальчиков. В совокупности мышечная дистрофия Дюшенна и мышечная дистрофия Беккера поражают 5 человек из 1000. Большинство больных страдают мышечной дистрофией Дюшенна.
Дефект гена, который приводит к мышечной дистрофии Дюшенна, отличается от дефекта гена, который вызывает мышечную дистрофию Беккера, но в возникновении обоих дефектов задействован один и тот же ген (ген дистрофина). Ген для обоих этих синдромов является рецессивным и находится на X-хромосоме. Таким образом, несмотря на то, что женщина может быть носителем дефектного гена, у нее не будет развиваться заболевание, потому что нормальный ген на одной X-хромосоме компенсирует дефект гена на другой Х-хромосоме. Однако у мужчины, который получает дефектный ген, это заболевание разовьется, потому что у мужчин только одна Х-хромосома ( Х-сцепленное наследование Х-сцепленное наследование Гены представляют собой сегменты дезоксирибонуклеиновой кислоты (ДНК) и содержат код для определенного белка, функционирующего в одном или нескольких типах клеток в организме. Хромосома состоит. Прочитайте дополнительные сведения ).
У мальчиков с мышечной дистрофией Дюшенна отсутствует почти весь мышечный белок дистрофин, который имеет большое значение для поддержания структуры мышечных клеток. У мальчиков с мышечной дистрофией Беккера дистрофин вырабатывается, но ввиду того, что структура белка изменена, дистрофин функционирует неправильно или количество дистрофина недостаточно.
Симптомы
Основным симптомом, который вызывает мышечная дистрофия Дюшенна и мышечная дистрофия Беккера, является слабость мышц, включая сердечную мышцу и дыхательные мышцы. Симптомы возникают только у мальчиков.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна начинается в возрасте от 2 до 3 лет. Первые симптомы — это задержка развития (особенно задержка в умении ходить) и трудности при ходьбе, беге, прыжках и подъеме по лестнице. Мальчики с мышечной дистрофией Дюшенна часто падают, что зачастую приводит к переломам рук или ног. Они ходят вразвалку, часто ходят на цыпочках и с трудом поднимаются с пола.
Затем обычно следует слабость в плечевых мышцах, которая постепенно ухудшается. Когда мышцы ослабляются, они также увеличиваются, но аномальная мышечная ткань не обладает силой. У мальчиков с мышечной дистрофией Дюшенна сердечная мышца также постепенно увеличивается и ослабляется, вызывая проблемы с сердцебиением. Осложнения со стороны сердца возникают примерно у одной трети мальчиков с мышечной дистрофией Дюшенна к возрасту 14 лет и у всех больных старше 18 лет. Однако ввиду того что эти мальчики не могут упражняться, ослабленная сердечная мышца не вызывает симптомов, пока заболевание не прогрессирует. Около одной трети больных мальчиков имеют умственное расстройство Ограничение умственных способностей Ограничение умственных способностей — это серьезное снижение интеллектуальной деятельности ниже среднего показателя, присутствующее с рождения или раннего младенческого возраста и приводящее. Прочитайте дополнительные сведения легкой степени, которое не ухудшается со временем и затрагивает главным образом речевые способности.
У мальчиков с мышечной дистрофией Дюшенна мышцы рук и ног обычно сокращаются вокруг суставов таким образом, что локти и колени не могут полностью разогнуться. Со временем развивается патологическое искривление позвоночника ( сколиоз Сколиоз Сколиоз — аномальное искривление позвоночника. Сколиоз может присутствовать при рождении или развиться в подростковом возрасте. Легкие формы могут вызывать только небольшой дискомфорт, но более. Прочитайте дополнительные сведенияМышечная дистрофия Беккера
У мальчиков с мышечной дистрофией Беккера слабость не такая выраженная и впервые появляется чуть позже, в возрасте около 12 лет. Обычно они способны ходить, по крайней мере, до 15 лет, и многие из них сохраняют способность ходить в зрелом возрасте. Характер слабости напоминает мышечную дистрофию Дюшенна. Однако очень немногие подростки нуждаются в инвалидной коляске. Большинство из них доживают до 30 или 40 лет.
Диагностика
Иногда биопсия мышц
Врачи выявляют мышечную дистрофию на основании характерных симптомов, например, мальчик становится слабым и растет слабее, особенно если в семейном анамнезе есть мышечная дистрофия или необъяснимая слабость у мальчиков. Врачи выполняют анализы крови, чтобы измерить уровень фермента креатинкиназы. При мышечной дистрофии креатинкиназа просачивается из мышечных клеток, что приводит к аномальному повышению ее уровня в крови. Однако высокий уровень креатинкиназы в крови не обязательно означает, что это мышечная дистрофия, потому что другие заболевания мышц также могут вызывать повышение уровня этого фермента. Врачи часто проводят исследование под названием электромиография Электромиография и исследования нервной проводимости Для подтверждения диагноза, предполагаемого на основании медицинского анамнеза и неврологического обследования, может понадобиться выполнение диагностических процедур. Электроэнцефалография. Прочитайте дополнительные сведенияДиагноз мышечной дистрофии Дюшенна ставится путем проведения генетических анализов (анализов ДНК) с использованием образца крови для выявления мутаций в гене дистрофина. Если генетические анализы не позволяют подтвердить диагноз, врачи проводят биопсию мышц (взятие кусочка мышечной ткани для исследования под микроскопом) для определения уровней белка дистрофина в мышце. Под микроскопом врачи видят мертвые ткани и аномально большие мышечные волокна. У людей с мышечной дистрофией Дюшенна белок дистрофина не обнаруживается совсем или обнаруживается в очень низких концентрациях.
Аналогичным образом, диагноз мышечной дистрофии Беккера ставится в случае, когда генетические анализы показывают наличие дефектов в гене дистрофина. Биопсия мышц показывает низкий уровень белка дистрофина в мышце, но не такой низкий, как при мышечной дистрофии Дюшенна.
Дети с мышечной дистрофией Дюшенна проходят электрокардиографию Электрокардиография Электрокардиография (ЭКГ) представляет собой быструю, простую и безболезненную процедуру, в ходе которой электрические импульсы сердца усиливаются и записываются. Эта запись, электрокардиограмма. Прочитайте дополнительные сведения для выявления проблем с сердцем. Эти исследования проводятся во время постановки детям диагноза или к 6-летнему возрасту.
Близкие родственники детей с мышечной дистрофией Дюшенна или Беккера могут пройти тесты для обнаружения гена. Пренатальная диагностика плода может помочь определить вероятность заболевания ребенка.
Лечение
Физиотерапия или ортезы для ног или лодыжек
Иногда ингибиторы ангиотензин-превращающего фермента и бета-блокаторы
Иногда препараты, увеличивающие выработку белка дистрофина
Иногда хирургическое вмешательство
При дистрофии Дюшенна — преднизон или дефлазакорт
Мышечные дистрофии Дюшенна и Беккера неизлечимы.
Дети с проблемами дыхания могут носить специальные маски, которые помогают дышать Альтернативные варианты Искусственная вентиляция легких — это использование аппарата для облегчения движения воздуха в легкие и из легких. Некоторые люди с дыхательной недостаточностью нуждаются в использовании аппарата. Прочитайте дополнительные сведения . Если маска не помогает нормально дышать, врачи могут вставить пластиковую трубку непосредственно в дыхательное горло (трахею) через небольшой разрез в передней части шеи (процедура называется трахеостомия). При необходимости для облегчения дыхания используется аппарат искусственной вентиляции легких Искусственная вентиляция легких Искусственная вентиляция легких — это использование аппарата для облегчения движения воздуха в легкие и из легких. Некоторые люди с дыхательной недостаточностью нуждаются в использовании аппарата. Прочитайте дополнительные сведения (устройство, помогающего воздуху попадать и выходить из легких). Трахеостомия может позволить детям с дистрофией Дюшенна прожить до третьего десятка. Детям с сердечными заболеваниями могут назначить лекарственные препараты, такие как ингибиторы ангиотензин-превращающего фермента и бета-блокаторы.
Детям с мышечной дистрофией Дюшенна, которым исполнилось 5 лет и у которых наблюдается значительная мышечная слабость, могут назначить преднизон или дефлазакорт, которые относятся к группе кортикостероидов. Преднизон или дефлазакорт принимаются внутрь ежедневно. При длительном приеме эти препараты приносят большую пользу, например, увеличивают силу, позволяя детям ходить еще несколько лет, сохраняют функцию сердца и легких и повышают выживаемость на 5–15 лет. Тем не менее, длительное применение этих препаратов вызывает много побочных эффектов, например, повышение массы тела, отечность лица и повышение риска развития проблем с позвоночником и костями ( Кортикостероиды Применение и побочные эффекты Кортикостероиды Применение и побочные эффекты ). Преднизон и дефлазакорт для лечения мышечной дистрофии Беккера не были достаточно изучены.
Некоторым людям с мышечной дистрофией Дюшенна и определенными мутациями в гене дистрофина могут быть назначены лекарственные препараты этеплирсен или голодирсен. Эти препараты помогают стимулировать выработку белка дистрофина. Хотя вырабатываемый дистрофин не является нормальным, он функционирует и может ослабить симптомы.
Дополнительная информация
Ниже приведен ресурс на английском языке, который может быть полезным. Обратите внимание, что составители СПРАВОЧНИКА не несут ответственности за содержание этого ресурса.
ПРИМЕЧАНИЕ: Это — пользовательская версия ВРАЧИ: Нажмите здесь, чтобы перейти к профессиональной версии
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Мышечная дистрофия Дюшенна/Беккера
Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма - существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.
DMD (DYSTROPHIN) - ген дистрофина, находится в Х-хромосоме в регионе Хр21.2 –р21.1, состоит из 79 экзонов. У 60%-70% больных выявляются крупные делеции, захватывающие один или несколько экзонов гена и локализованные в двух "горячих" регионах - в области 5" конца (экзоны 6-19) и 3" конца (экзоны 40-43). У 5% больных обнаруживаются дупликации, в остальных случаях - точковые мутации. Различия в тяжести клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в характере мутации в гене дистрофина. При мышечной дистрофии Дюшенна мутации в гене дистрофина приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При мышечной дистрофии Беккера структурные перестройки гена не приводят к сдвигу рамки считывания, ДНК-полимераза может "перескакивать" делетированные экзоны, что приводит к синтезу внутренне усеченного белка, который может, до некоторой степени, выполнять свои функции.
Нейромышечное заболевание, обусловленное мутацией в гене дистрофина и приводящее к прогрессирующей дегенерации мышечных волокон.
Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных волокон и замещение их соединительнотканными структурами, которые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии. Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.
Заболевание проявляется в возрасте 1-5 лет, быстро прогрессирует и приводит к летальному исходу до 25 летнего возраста. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук, могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-ти летнего возраста, после чего пользуются инвалидной коляской. Гибель больных наступает от сердечной недостаточности или от интеркуррентных инфекций.
Наиболее часто заболевание возникает в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног. Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилятационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.
Описаны клинические проявления ПМДД у лиц женского пола, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс). Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Литература
Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Типичные клинические проявления.
При выявлении у ребенка - мать, братьев и сестер (в некоторых случаях сестер матери, двоюродных братьев и сестер со стороны матери и других родственников по рекомендации врача-генетика).
Литература
Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
- Мутация не выявлена
- Мутация выявлена в гемизиготном состоянии
Литература
Monaco, A. P.; Kunkel, L. M.: A giant locus for the Duchenne and Becker muscular dystrophy gene. Trends Genet. 3: 33-37, 1987.
Becker, P. E.: Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat. Neurol. Scand. 193: 427, 1955.
Прогрессирующая мышечная дистрофия Дюшенна — Беккера. Трудности диагностики
Цель статьи: представить клинический случай прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера и показать трудности и особенности диагностики этой генетической патологии.
Основные положения. В статье представлен анализ клинического случая диагностики ПМД Дюшенна — Беккера у мальчика 2 лет. Описаны этапы диагностического поиска, проведен анализ результатов клинического наблюдения и молекулярно-генетического обследования. Предложен алгоритм обследования при повышении активности аспартатаминотрасферазы (АСТ), аланинаминотрансферазы (АЛТ) неясного генеза.
Заключение. Описание клинического примера ПМД демонстрирует сложности, с которыми встречаются врачи разных специальностей при диагностике данного заболевания. Ранние клинические признаки, повышение в крови активности АСТ, АЛТ, креатинфосфокиназы являются основанием для назначения молекулярно-генетического исследования. Данную схему можно использовать при постановке диагноза ПМД, чтобы сократить длительные поиски несуществующей неврологической и инфекционной патологии, своевременно назначить адекватную терапию и улучшить качество жизни больного ребенка.
Вклад авторов: Царькова С.А. — проверка критически важного содержания, утверждение рукописи для публикации; Ушакова Р.А. — наблюдение, обследование пациента, сбор клинического материала, анализ и интерпретация данных; Громада Н.Е. — разработка концепции статьи, анализ и интерпретация данных, написание текста рукописи; Косенкова М.И., Мусалова О.Р. — обзор публикаций по теме статьи, написание текста рукописи.
Конфликт интересов: авторы заявляют об отсутствии возможных конфликтов интересов.
По данным Регистра врожденной и наследственной патологии Свердловской области клинико-диагностического центра «Охрана здоровья матери и ребенка», ежегодно выявляют 4–5 случаев прогрессирующей мышечной дистрофии (ПМД). Это труднокурабельное заболевание имеет высокую социальную значимость в связи с ранней инвалидизацией ребенка и необходимостью оказания своевременной психосоциальной помощи родителям и пациенту. Недооценка ранних симптомов ПМД Дюшенна — Беккера, клинических и нейрофизиологических критериев сопряжены с поздней диагностикой заболевания.
ПМД Дюшенна — Беккера — наследственное рецессивное нервно-мышечное заболевание, сцепленное с Х-хромосомой, вызванное мутациями в гене DMD , приводящими к отсутствию или недостаточной функции дистрофина, цитоскелетного белка, который обеспечивает прочность, стабильность и функциональность миофибрилл. Шифруется по классификации МКБ-10 как G71.0 1 .
Ген, отвечающий за выработку белка дистрофина, находится на Х-хромосоме (локализация Хр 21.2) и состоит из 79 частей-экзонов. При наличии мутаций в этом гене белок дистрофин не синтезируется, мышечная ткань гибнет, замещается жировой и соединительной тканью. В 40–60% случаев отмечается мутация (делеция — потеря или дупликация — удвоение) одного или нескольких экзонов [3, 4] .
Выделяют два клинических варианта: миодистрофии Дюшенна и Беккера. Миодистрофия Беккера (1 на 30 000 населения) — более легкий вариант заболевания, при котором синтез белка дистрофина идет не до конца, и в результате получается немного укороченный, но вполне функциональный белок. В данном случае болезнь протекает с медленным прогрессированием мышечной слабости и с сохранением способности к самостоятельной ходьбе в течение 15–20 лет от начала заболевания [3, 4] .
С миопатией Дюшенна рождается один из 5000 мальчиков (3,3 : 100 000 населения). Манифестация болезни чаще всего наблюдается в возрасте от 1 года до 5 лет. Заболевание характеризуется прогрессирующим злокачественным течением: формированием атрофии мышц тазового и плечевого пояса на фоне псевдогипертрофии икроножных, ягодичных, дельтовидных мышц, мышц живота и языка. Возможно снижение ментальной функции. Постепенно развивается деформация стоп, грудной клетки, позвоночника, прогрессируют дилатационная миокардиопатия и дыхательные нарушения, приводящие к летальному исходу в молодом возрасте [3, 4] .
Диагностические критерии заболевания следующие: пол пациента — мужской; установленный диагноз прогрессирующей миодистрофии у родственников мужского пола по материнской линии или неуточненное нервно-мышечное заболевание; установленный факт наличия в семье женщин-носительниц патологического гена; кардиологические заболевания (кардиомиопатии) у родственников женского пола по материнской линии; задержка становления двигательных навыков; снижение интеллекта; повышение активности трансаминаз АЛТ и АСТ и креатинфосфокиназы (КФК) в сыворотке крови.
Ранними симптомами заболевания являются повышенная утомляемость, мышечная слабость, гипотония в конечностях, псевдогипертрофия мышц голеней и бедер, частые падения или неуклюжесть, затруднение при приседании, беге, подъеме по лестнице, неспособность прыгать, использование вспомогательных приемов при подъеме с пола (ребенок помогает себе руками при подъеме из положения лежа, сидя), ходьба на носочках, задержка формирования речи [3, 4] .
В биохимическом анализе крови регистрируется повышение активности трансаминаз АЛТ, АСТ, лактатдегидрогеназы, КФК [5] .
При проведении электромиографии определяется миопатический тип с уменьшением (укорочением) величины средней длительности потенциала двигательных единиц (ПДЕ), снижением амплитуды отдельных ПДЕ.
На УЗИ мышц появляются признаки замены мышечной ткани жировой или фиброзной тканью. По данным ЭКГ, возможно появление аритмии, нарушение проводимости, на ЭхоКГ — признаки систолической дисфункции, дилатации левого желудочка, гипертрофии миокарда, митральной регургитации.
Для выявления дегенерации мышечной ткани используют МРТ и магнитно-резонансную спектроскопию мышц голени, бедер, таза (иногда денситометрию) [5] .
Генетические исследования MLPA (Multiplex Ligation-dependent Probe Amplification), секвенирование гена DMD предполагают проведение сравнительной геномной гибридизации массива и поиск точечных мутаций [6] .
В настоящее время лечение — симптоматическое, направленное на улучшение качества жизни. Применение ГКС позволяет замедлить прогрессирующую атрофию мышечной ткани. Препаратами выбора являются преднизолон или дефлазакорт. Назначение ингибиторов АПФ или β-блокаторов рекомендуют для профилактики дилатационной кардиомиопатии, мочегонных препаратов — при наличии сердечно-сосудистой недостаточности. Для профилактики остеопороза назначают препараты, содержащие витамин D2 и кальций [7] .
Детям с данной патологией рекомендуется физиотерапия, направленная на поддержание физической активности: профилактические растяжки, применение ортопедических аппаратов, лечебная физкультура [3] .
На стадии клинического эксперимента находится метод экзон-скиппинга — пропуска поврежденных экзонов, который предполагает «достройку» обходного параллельного пути для рамки считывания, минуя поврежденный экзон, что приводит к синтезу укороченного белка, сохраняющего свою функциональность.
Генно-клеточная терапия также находится в процессе разработки: использование стволовых клеток, «уснувшего» гена — утрофина (эмбрионального дистрофина), доставка в клетки генных конструкций микрогенов с помощью аденоассоциированных вирусов [8] .
Пациент находился под наблюдением в отделении патологии детей раннего возраста № 2 МАУ «Детская городская клиническая больница № 11» г. Екатеринбурга. У родителей ребенка получено добровольное информированное согласие на публикацию данных.
В результате ретроспективного анализа медицинской документации (индивидуальная карта развития ребенка Ф112/У) и опроса родителей выявлено, что мальчик рожден женщиной 22 лет от 1-й физиологически протекавшей беременности, в результате самопроизвольных срочных родов в головном предлежании плода на 39 неделе гестации. Масса тела ребенка при рождении — 3260 г, длина тела — 53 см, окружность головы — 36 см, окружность груди — 34 см, что соответствовало сроку гестации. Оценка по шкале Апгар — 8/9 баллов. Интранатальный и неонатальный периоды — без особенностей. Результат неонатального скрининга на наличие врожденных заболеваний отрицательный.
Наследственный анамнез не отягощен. Выписан из роддома на 3-и сутки домой. Вакцинация проведена согласно национальному календарю. Находился на естественном вскармливании до 9 месяцев.
Ребенок в течение первого года жизни рос и развивался соответственно возрасту: начал держать голову в 1 месяц, следить за игрушкой в горизонтальной плоскости, гулить — с 2 месяцев, брать предметы в руки — в 3,5–4 месяца, поворачиваться со спины на живот — в 5,5 месяцев, сидеть без поддержки — в 6 месяцев, ползать — с 9 месяцев, произносить слово «мама», стоять с опорой — в 12 месяцев, ходить самостоятельно — с 14 месяцев.
В возрасте 18 месяцев у ребенка выявлена гепатоспленомегалия (печень выступала из-под края реберной дуги на 3,5 см, селезенка — на 2 см), по данным УЗИ органов брюшной полости: правая доля печени — 74,5 мм, левая доля — 38 мм, длина селезенки — 78 мм, толщина — 34,2 мм. На фоне отсутствия синдрома желтухи и при нормативных показателях билирубина в анализе крови обнаружено стойкое повышение (в течение 6 месяцев) активности трансаминаз: АСТ — 360–390–426 Е/л, АЛТ — 298–417 Е/л (при норме до 45 Е/л и 40 Е/л соответственно), γ-глютамилтранспептидазы — 32,4 Е/л (при норме 6–23 Е/л). Пациент проконсультирован врачом иммунологом-инфекционистом. Методы ИФА и ПЦР-диагностики позволили исключить гепатит инфекционной этиологии.
На основании сведений, полученных от родителей, установлено, что после 16 месяцев жизни у мальчика появились скованность в ногах по утрам и неустойчивость при ходьбе, частые падения.
К 24 месяцам жизни физическое развитие по уровню биологической зрелости соответствовало паспортному возрасту. Масса — 14,5 кг. Рост — 93 см. Окружность головы — 48 см. Телосложение пропорциональное. Кожа эластичная, чистая. Сознание ясное. Очаговых и менингеальных симптомов нет. Чувствительная сфера и черепные нервы — без патологии.
Снижен мышечный тонус в нижних конечностях. Биципитальные и карпорадиальные, коленные и ахилловы рефлексы снижены и симметричны. Мышечная сила в верхних и нижних конечностях снижена до 4 баллов (определение по принципу «напряжения — преодоления»). Псевдогипертрофия икроножных мышц. Походка неуверенная. Позитивный симптом Говерса: поднимается из положения лежа и на корточках, опираясь руками о пол и колени. Сидит и опирается на руку. Бег затруднен. Не подпрыгивает. Частично понимает речь, выполняет инструкции матери. Произносит отдельные слова.
Функции слуха и зрения сохранены. Функции тазовых органов не нарушены.
Используя скрининговые центильные графики подуровней нервно-психического развития для индивидуальной оценки тестируемых навыков и умений [9], мы определили, что подуровень ручной умелости, развития речи и социальной адаптации находится в пределах 25–75% центильной зоны и соответствует возрасту. Подуровень общей моторики по тестируемым навыкам и поздние сроки их появления находятся за пределами 90% центильной зоны и свидетельствуют о задержке и дисгармоничном развитии.
Со стороны органов дыхания и сердечно-сосудистой системы отклонений нет. Данные ЭКГ и ЭхоКГ — без патологии. Результаты электронейромиографии: аксональная нейропатия малоберцового нерва слева.
С учетом описанного выше неврологического дефицита, пола пациента, признаков синдрома цитолиза, стойкого увеличения активности трансаминаз на фоне отсутствия желтушного синдрома решено провести исследование уровня КФК в крови. Выявлено превышение показателя в 70 раз — 17 453 Е/л (при референсных значениях до 247 Е/л). Это стало показанием для поиска заболеваний, ассоциированных с наследственными формами ПМД.
Проведено молекулярное-генетическое исследование. На первом этапе делеции и дупликации в гене DMD методом MLPA не обнаружены. Затем с помощью молекулярно-генетического метода массового параллельного секвенирования найден вариант в гемизиготном состоянии в 13 экзоне гена DMD (chrX:32613972 G>A, c.1504C>T) (точечная мутация). Описан ранее в литературе как патогенный. Валидирован методом прямого секвенирования по Сэнгеру.
Тип наследования — X-сцепленный рецессивный. Генетический риск для сибсов: по заболеванию для братьев — до 50%, по носительству для сестер — 50%. Для уточнения риска необходимо проведение молекулярно-генетической диагностики у матери для поиска выявленной мутации в гене DMD .
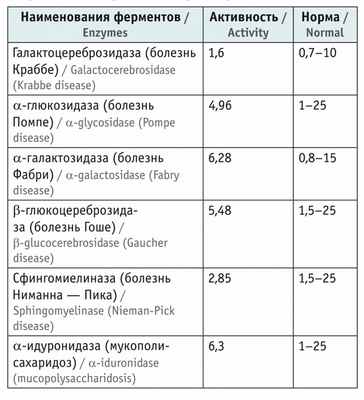
Активность лизосомных ферментов в крови у пациента находилась в пределах референсных значений ( табл. ).
Показатели активности лизосомных ферментов в пятнах высушенной крови, мкМ/л/ч
Ребенку назначено лечение: преднизолон в дозе 0,5 мг/кг/сутки. Рекомендована физиотерапия с дозированной физической нагрузкой. Предложена социально-психологическая помощь родителям: работа с психологом, даны координаты Благотворительного фонда помощи детям с миодистрофией Дюшенна и иными тяжелыми нервно-мышечными заболеваниями «МойМио».
Из представленных данных следует, что ребенку в возрасте 2 лет на основании жалоб, данных анамнеза, особенностей неврологического статуса, результатов лабораторно-инструментальных и молекулярно-генетических исследований был поставлен диагноз: мышечная дистрофия Дюшенна — Беккера, стадия сохраненной способности к самостоятельному перемещению. Проведение энзимодиагностики позволило исключить некоторые наследственные лизосомные болезни накопления (см . табл. ).
У пациента дебют заболевания наступил в возрасте 2 лет. Минимальные клинические признаки ПМД остались незамеченными, родители первыми обратили внимание на нарастание мышечной слабости после 16–18 месяцев, что явилось поводом обращения к педиатру и неврологу. Симптомы дистрофии прогрессировали медленно с неравномерным поражением мышц, в дебюте заболевания пострадали отдельные группы мышц, что привело к относительной компенсации двигательных расстройств.
По нашим и литературным данным, в структуре ошибочного диагноза доминируют перинатальная энцефалопатия, гепатит, гепатоз, кардиомиопатия, суставно-мышечная патология [3, 4] . У наблюдаемого ребенка отмечались стойкое длительное (в течение 6 месяцев) увеличение активности трансаминаз, наличие гепатоспленомегалии, что заставило исключить гепатит инфекционной этиологии. Высокие показатели КФК обнаружили позже.
В данном случае, при миодистрофии Дюшенна, повышение активности АЛТ и АСТ в анализах крови имеет внепеченочное происхождение и определяется при разрушении миофибрилл мышц на фоне высоких показателей КФК.
Известно, что алгоритм диагностики ПМД состоит из нескольких этапов. Если ребенок начинает ходить поздно, выявлен положительный симптом Говерса или другие нарушения мышечной функции, имеется отягощенный семейный анамнез, в крови — увеличение активности трансаминаз неуточненного генеза, то рекомендуется определение уровня КФК. В свою очередь, высокие показатели КФК создают необходимость проведения молекулярно-генетической диагностики мутации гена дистрофина и биопсии мышц ребенка [3] .
Для структурирования диагностического поиска заболевания мы использовали адаптированный алгоритм Р.А. Ушаковой [10] . Согласно представленной схеме, у пациента наблюдалось повышение активности трансаминаз неясного генеза, положительный симптом Говерса, отсутствие ПМД в семейном анамнезе ( рис. ).
Рис. Алгоритм диагностики прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера [10]
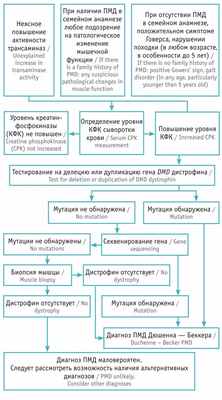
Для верификации диагноза прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера при наличии у пациента клинической симптоматики, характерной для данного заболевания, необходимо проведение молекулярно-генетического исследования с целью поиска мутации в гене DMD . Аналогичное обследование предлагают матери пациента для определения типа наследования, расчета риска патологии при планировании следующей беременности.
Данную схему можно использовать при постановке диагноза ПМД даже при отсутствии пренатальной диагностики, указаний на заболевание в семейном анамнезе, чтобы сократить длительные поиски несуществующей неврологической и инфекционной патологии, обратить внимание на ранние признаки заболевания, своевременно провести определение уровня креатинфосфокиназы, молекулярно-генетическое исследование, назначить адекватную терапию и улучшить качество жизни больного ребенка.
Мышечная дистрофия, или миодистрофия
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Мышечная дистрофия: причины появления, симптомы, диагностика и способы лечения.
Определение
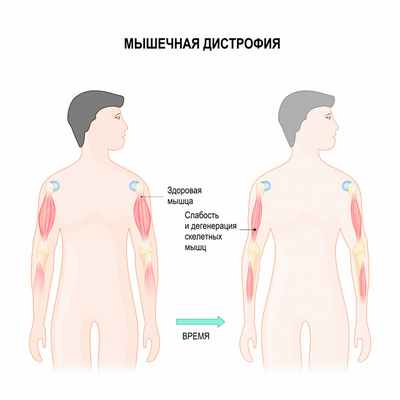
Мышечные дистрофии - это большая группа наследственных заболеваний, которые характеризуются прогрессирующей слабостью и дегенерацией скелетных мышц, то есть потерей мышечной массы. К наиболее распространенным миодистрофиям относятся миодистрофия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия), мышечная дистрофия Дюшенна и дистрофия Беккера. Также в эту группу входят дистрофия Эмери-Дрейфуса, миодистрофия Эрба-Рота и другие.
Миодистрофия Дюшенна поражает в основном мальчиков, а распространенность этого заболевания составляет 3,3 на 100 000 населения, 1 из 3500 новорожденных мальчиков страдает данной патологией. Этот вид дистрофии часто выделяют в одну группу с миодистрофией Беккера (частота встречаемости 1 на 20 000 новорожденных). Миодистрофии Дюшенна и Беккера наследуются по Х-сцепленному рецессивному типу. Это означает, что повреждение находится в Х-половой хромосоме и передается от матери к сыну, а дочери являются носительницами и, как правило, сами не болеют.
Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей.
Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них - Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
Частота встречаемости миодистрофии Эрба-Рота составляет от 1,5 до 2,5 случаев на 100 тыс. населения. Этому типу мышечной дистрофии подвержены и мальчики, и девочки. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно, то есть патология проявляется, если ребенок получает аномальный ген от каждого из родителей. Каждый родитель может быть носителем дефектного гена, но обычно остается здоровым. Около 30% генных мутаций возникают de novo, то есть не наследуются, а появляются «ниоткуда» и далее могут передаваться потомству.
Причины появления мышечной дистрофии
Причиной развития разных миодистрофий являются патологии в генах - известно порядка 25 генов, ответственных за развитие врожденных миодистрофий.
При мышечной дистрофии Дюшенна вследствие мутации нарушается выработка белка дистрофина, который обеспечивает прочность, стабильность и функциональность мышечных волокон, и его нехватка приводит к повреждению мембран мышечных клеток (миоцитов).
Классификация заболеваний
Мышечные дистрофии могут классифицироваться в зависимости от того, какой белок подвергся мутации. Кроме того, их подразделяют по типу наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные.
Симптомы мышечной дистрофии
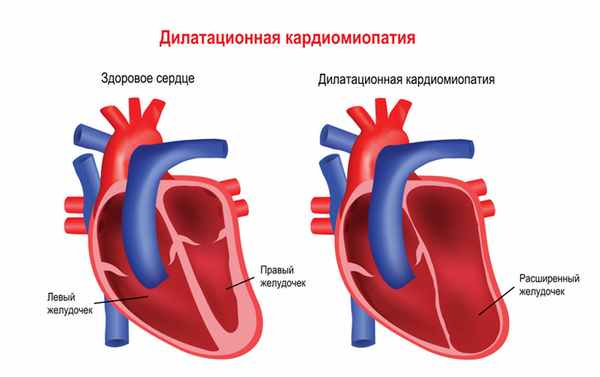
Мышечная дистрофия Дюшенна обычно манифестирует в возрасте 2-3 лет. Патологические процессы сначала происходят в мышцах ног, дети могут ходить на пальцах, вразвалку, отмечается избыточное выгибание позвоночника вперед – лордоз. Детям становится сложно бегать, прыгать, подниматься по лестнице, вставать с пола. Для разных типов миодистрофий характерным является симптом Говерса – вследствие слабости мышц бедер и тазового пояса больному, чтобы подняться из положения на корточках, приходится опираться руками об пол, затем подниматься, опираясь руками об колени. Мышечная слабость прогрессирует, у детей развивается сколиоз и сгибательные контрактуры – когда ребенок не может полностью разогнуть конечность. Дети часто падают, поэтому велик риск переломов рук или ног. Отдельные мышечные группы могут замещаться жировой или фиброзной тканью, в результате чего появляется псевдогипертрофия мышц, особенно заметная на лодыжках. Если страдает миокард (сердечная мышца), то существует предрасположенность к развитию нарушений ритма и проводимости сердца, а также дилатационной кардимиопатии (состояния, когда камеры сердца увеличены, а стенки истончены), приводящей к сердечной недостаточности.
В 20-30% случаев при мышечной дистрофии Дюшенна появляются нарушения интеллекта и памяти.
К 12 годам большинство детей вынуждено пользоваться инвалидной коляской. В возрасте 15-20 лет пациентам уже требуется респираторная поддержка, умирают больные миодистрофией Дюшенна от дыхательных или кардиальных осложнений в возрасте 12-25 лет.
Миодистрофия Беккера дебютирует в 10-20 лет и медленно прогрессирует, способность к самостоятельной ходьбе сохраняется в течение 15-20 лет от начала заболевания. Симптоматика схожа с миодистрофией Дюшенна, слабость распространяется на мышцы бедер, таза, плеч, пациенты ходят на носочках или вразвалку, также наблюдается гипертрофия мышц голеней.
Развитие заболевания очень индивидуально, некоторым пациентам требуется инвалидное кресло к 30 годам, некоторые длительное время обходятся тростью.
У пациентов с миодистрофией Беккера также отмечается поражение сердечной мышцы с развитием сердечной недостаточности.
Первые признаки миодистрофии Ландузи-Дежерина проявляются в основном в возрасте 10-20 лет. Сначала атрофия и мышечная слабость наблюдаются в плечевом поясе с поражением мышц лопаток и плеч, потом распространяются на лицо с характерной асимметричностью. Начальными проявлениями являются затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании заболевания страдают лицевые мышцы, при этом пациент не может крепко зажмурить глаза и сжать губы. Позже мимика становится скудной, а речь неразборчивой. Характерными симптомами являются поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира»), «полированный» лоб. Иногда атрофия распространяется на мышцы ног. Другими клиническими признаками миодистрофии Ландузи-Дежерина могут быть аномалии сосудов сетчатки глаза, отек и отслойка сетчатки, снижение слуха.
Для миодистрофии Эмери-Дрейфуса характерны контрактуры локтевых и голеностопных суставов, возникающие в раннем детстве (укорочение ахилловых сухожилий приводит к тому, что ребенок не может опуститься на пятки), тугоподвижность позвоночника, медленно прогрессирующая слабость лопаточно-плечевых и тазово-перонеальных мышц (мышц бедра и голени), а также выраженная кардиомиопатия с нарушениями ритма и проводимости. Тяжесть заболеваний сердца часто определяет прогноз течения болезни вследствие высокой вероятности внезапной сердечной смерти или развития прогрессирующей сердечной недостаточности.
Миодистрофия Эрба-Рота сопровождается слабостью мышц поясничной области и конечностей. Первые признаки появляются в возрасте 10-20 лет: трудности при беге, быстрой ходьбе, прыжках, характерен симптом Говерса. Со временем начинает меняться осанка, походка, снижается тонус мышц плечевого пояса. С прогрессированием заболевания больной может полностью потерять способность ходить. Тотальная гипотрофия мышц туловища приводит к тому, что у пациента начинают выступать лопатки, талия становится очень тонкой, усиливается поясничный лордоз. Характерен симптом свободных надплечий — при попытке приподнять больного, удерживая его подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними.
Диагностика мышечной дистрофии
Предварительный диагноз врач может установить уже при осмотре, наблюдая за попытками ребенка побежать, прыгнуть, подняться по ступенькам, встать с пола.
Для подтверждения диагноза проводятся:
- Анализ крови на сывороточную креатинкиназу, АСТ, АЛТ.
Публикации в СМИ
Мышечная дистрофия Дюшенна — наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением; наследуется по рецессивному X-сцепленному типу. Вариант мышечной дистрофии Дюшенна — мышечная дистрофия Беккера — имеет более доброкачественное течение.
Генетические аспекты • Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна–Беккера, *310200, Xp21.2, ген DMD дистрофина, À рецессивное) — возникает в результате дефектов гена, кодирующего белок дистрофин • Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов • Преобладающий пол — мужской, тем не менее мышечные дистрофии Дюшенна и Беккера могут встречаться у девочек при кариотипе X0, мозаицизмах X0/XX, X0/XXX и структурных аномалиях хромосом.
Патоморфология • Дистрофия мышечных волокон, первично-мышечный тип поражения • Фиброзные изменения в мышечных пучках • Местная воспалительная реакция.
Клиническая картина
• Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно со слабости мышц тазового пояса.
• Уже на первом году жизни отмечают отставание в психомоторном развитии. Больные дети позднее начинают садиться, вставать, ходить.
• Постепенно развиваются слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу утиной. Из горизонтального положения дети встают поэтапно с использованием рук (взбирание лесенкой).
• Отмечаются симметричные атрофии проксимальных групп мышц нижних конечностей (мышцы таза и бедра). Атрофия через 1–3 года распространяется на проксимальные группы мышц верхних конечностей.
• Атрофии мышц приводят к развитию лордоза, крыловидных лопаток, осиной талии.
• Характерна псевдогипертрофия икроножных мышц.
• Мышцы при пальпации плотные, безболезненные.
• Мышечный тонус обычно снижен в проксимальных группах мышц.
• Изменения рефлексов •• Коленные рефлексы исчезают на ранних стадиях заболевания •• Позднее исчезают рефлексы с двуглавой и трёхглавой мышц плеча •• Ахилловы рефлексы обычно длительное время остаются сохранными.
• Дистальная мускулатура конечностей поражается на поздних стадиях заболевания.
• Костно-суставные нарушения — деформации позвоночника, стоп, грудной клетки; рентгенологически обнаруживают сужение костномозгового канала, истончение коркового слоя диафизов длинных трубчатых костей.
• Сердечно-сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность, изменения на ЭКГ.
• Нейроэндокринные нарушения выявляют у 30–50% больных — синдром Иценко–Кушинга, адипозогенитальная дистрофия.
• Психические нарушения — олигофрения в форме дебильности или имбецильности.
• Клинические проявления мышечной дистрофии Беккера обычно начинаются в 10–15 лет. От мышечной дистрофии Дюшенна отличается доброкачественным течением и более поздним возникновением тяжёлых симптомов. Сухожильные рефлексы долгое время остаются сохранными. Поражения внутренних органов менее выражены, интеллект сохранён.
Лабораторные исследования. Для мышечной дистрофии Дюшенна типично раннее (с 5 дня жизни) увеличение активности КФК в крови (в 30–50 раз выше нормы).
Дифференциальная диагностика. Мышечную дистрофию Дюшенна–Беккера дифференцируют от других мышечных дистрофий, рахита, врождённого вывиха бедра.
ЛЕЧЕНИЕ
Режим амбулаторный с наблюдением у невропатолога, хирурга-ортопеда, терапевта и профпатолога, работника социальной сферы и протезиста.
Мероприятия • Лечение мышечной дистрофии Дюшенна направлено на поддержании физической активности пациента и улучшение качества его жизни; как правило, быстро становится неэффективным • Физические упражнения выполняют систематически и по определённой схеме. Короткие перерывы показаны при возникновении болей в мышцах и мышечной усталости • Использование протезов позволяет больным двигаться и замедляет формирование сколиоза • Поддержание дыхания, ИВЛ во время сна для предотвращения синдрома ночной гиповентиляции • Экспериментальные методы, в особенности генная терапия (гены дистрофина и утрофина), чрезвычайно перспективны, хотя и не получили пока клинического распространения.
Оперативное лечение. Ортопедическое вмешательство необходимо при наличии контрактур и фиксации суставов.
Лекарственная терапия • ГК (преднизолон по 0,75 мг/кг/сут) увеличивают мышечную силу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование заболевания • При длительной стероидной терапии необходим тщательный контроль развития побочных эффектов, включающий наблюдение за массой тела, АД, состоянием слизистой оболочки ЖКТ и иммунной системы.
Наблюдение. Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни пациентов.
Течение и прогноз • Течение мышечной дистрофии Дюшенна быстропрогрессирующее, злокачественное • Значительные двигательные расстройства, развивающиеся ко второму десятилетию жизни, ограничивают самостоятельное передвижение больных • Смерть наступает на втором или третьем десятилетии жизни, часто в результате пневмонии • Течение мышечной дистрофии Беккера медленнопрогрессирующее. Больные длительное время сохраняют работоспособность.
Профилактика состоит в генетическом консультировании.
Синонимы • Прогрессирующая мышечная дистрофия Дюшенна • Псевдогипертрофическая мышечная дистрофия Дюшенна • Дистрофия Дюшенна • Болезнь Дюшенна • Миопатия псевдогипертрофическая • Миопатия псевдогипертрофическая Дюшенна.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Примечания • Термин «псевдогипертрофическая прогрессирующая мышечная дистрофия» объединяет мышечные дистрофии Дюшенна и Беккера • Мышечная дистрофия Дюшенна описана в 1853 г. Дюшенном • Мышечная дистрофия Беккера описана в 1955 г. Беккером.
Читайте также:
- Паразитизм. Антагонистический симбиоз. Факультативные паразиты. Облигатные паразиты. Метабиоз. Сателлизм. Антагонизм.
- Эпидемиология внутрибольничных инфекций. Распространение внутрибольничных инфекций.
- Стенки живота. Переднебоковая стенка живота. Топография переднебоковой стенки живота.
- Ревматическая лихорадка
- Врожденный локализованный фиброматоз. Фиброматоз шеи.