Надпочечниковая вирилизация
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Нюансы определения пола при адреногенитальном синдроме
По данным неонатальных скрининговых программ разных стран, популяционная распространенность сольтеряющей формы адреногенитального синдрома (АГС), когда вырабатывается избыток андрогенов, но мало глюкокортикоидов и минералокортикоидов, варьирует от 1 на 10 000 до 1 на 15 000 детей
Адреногенитальный синдром
По данным неонатальных скрининговых программ разных стран, популяционная распространенность сольтеряющей формы адреногенитального синдрома (АГС), когда вырабатывается избыток андрогенов, но мало глюкокортикоидов и минералокортикоидов, варьирует от 1 на 10 000 до 1 на 15 000 детей.
Высокая распространенность АГС, серьезные медицинские и социально-экономические последствия несвоевременной диагностики, наличие специфического лабораторного маркера — 17-ОНП (17-гидрокси-прогестерона) — обусловили внедрение массового неонатального скрининга во многих странах, в т. ч. в США, Франции, Германии, Италии, Швейцарии, Японии, Швеции, Индии и др. Он позволяет раньше выявить болезнь, правильно определить половую принадлежность ребенка и зарегистрировать паспортный пол, предотвратить развитие острого надпочечникового криза и летальность. Скрининг — единственно возможный метод доклинической диагностики вириальной формы болезни; в Беларуси он еще не используется.
Задача врача — установить пол, не ограничиваясь беглым осмотром; предупредить психологические проблемы и дополнительные терапевтические вмешательства, связанные с необходимостью перемены пола и выраженной низкорослостью пациента; исключить неоправданное назначение глюкокортикоидов.
Адреногенитальный синдром — это группа наследственных заболеваний с аутосомно-рецессивным типом наследования, имеющих клинические и метаболические проявления. В основе нарушений — дефекты ферментов, участвующих в биосинтезе гормона надпочечников — кортизола. Наиболее распространенная форма АГС (90–95% случаев) — дефицит фермента 21-гидроксилазы (21-ГД), который участвует в промежуточном этапе биосинтеза кортикостероидов. Из-за дефицита этого фермента надпочечники недостаточно вырабатывают кортизол и альдостерон. В крови больного значительно увеличивается уровень стероидов и андрогенов. Стабильное повышение концентрации последних отмечается у плода с 7-й недели беременности. У девочек наблюдается внутриутробная вирилизация (появление вторичных половых признаков по мужскому типу), у мальчиков — клиническая картина ложного преждевременного полового развития.
При рождении у девочек отмечается гипертрофия клитора, сращение мошоночного шва, формирование урогенитального синуса. В редких случаях внутриутробная андрогенизация выражена настолько, что наружные половые органы ребенка имеют полностью мужское строение (ложный женский гермафродитизм). При УЗИ органов малого таза видны внутренние гениталии (матка, фаллопиевы трубы и яичники).
Для исключения ложного женского гермафродитизма все новорожденные с фенотипическим мужским полом и не пальпируемыми в мошонке и по ходу паховых каналов яичками, а также дети, у которых бисексуальное строение гениталий, должны обследоваться с обязательным кариотипированием. Кариотип 46ХХ при гермафродитных наружных половых органах, высокий для данного срока гестации и массы тела ребенка уровень 17-ОНП, наличие матки при УЗИ с 95% вероятностью свидетельствуют об АГС у девочек.
Повышенный уровень надпочечниковых андрогенов у мальчиков внутриутробно не имеет принципиального значения, т. к. яички начиная с 1–2-го триместра беременности активно вырабатывают тестостерон. При рождении наружные половые органы сформированы правильно, соответственно мужскому типу. Часто наблюдается пигментация мошонки и сосков.
С возрастом избыток андрогенов надпочечников приводит к увеличению размеров клитора у девочек или полового члена у мальчиков, появлению оволосения с 2–3 лет, кратковременному ускорению физического развития (до 9–10 лет), быстрому закрытию зон роста и выраженной низкорослости ребенка. Окончательный рост пациентов с адреногенитальным синдромом ниже генетического на 1–2 стандартных отклонения.
При полной потере активности фермента 21-ГД в периоде новорожденности наблюдается недостаточность минералокортикоидов, развивается тяжелая сольтеряющая форма заболевания. Она проявляется кризом. На 2–3-й неделе жизни у ребенка появляются неспецифические симптомы: вялое сосание, частые срыгивания и рвота, диарея, потеря массы тела. Нарастание электролитного дисбаланса приводит к развитию гипонатриемической дегидратации, гиперкалиемии, метаболическому ацидозу и кардиогенному шоку. Эти клинико-биохимические нарушения бывают при многих состояниях периода новорожденности и связаны с гастроинтестинальными (гастроэнтерит, пилоростеноз) и ренальными (острый пиелонефрит, почечная дисплазия) потерями, патологией надпочечников (врожденными гипоплазией, псевдо- и гипоальдостеронизмом).
Неправильная или поздняя диагностика адреногенитального синдрома (АГС), отсутствие адекватной заместительной гормональной терапии приводят к гибели ребенка в неонатальном периоде.
Для лабораторной диагностики этой формы АГС есть надежный патогенетический маркер — уровень 17-ОНП в крови. Определение повышенных концентраций биохимического показателя у новорожденных помогает выявить заболевание до начала клинической манифестации. Исходный уровень 17-ОНП примерно в 100 раз превышает нормальные показатели для возраста и массы тела ребенка.+
У недоношенных и детей с родовыми травмами (или рожденных с низким весом) при нормальных сроках гестации концентрации 17-ОНП могут быть повышенными и при отсутствии дефицита этого фермента. В таких случаях рекомендуется повторное определение гормонального показателя через 5–7 дней. Снижение уровней 17-ОНП в динамике позволяет исключить 21-гидроксилазную недостаточность.
Раннее выявление гормональных нарушений, характерных для адреногенитального синдрома, делает обязательным обследование всех новорожденных с гермафродитными гениталиями и мальчиков с синдромом потери соли. У последних отправные моменты диагностического поиска — отсутствие увеличения массы тела, клинические и лабораторные признаки (гипонатриемия, гиперкалиемия, метаболический ацидоз) сольтеряющего компонента. Изолированная вирильная форма заболевания у мальчиков диагностируется только к 4–5 годам при манифестации признаков преждевременного полового развития.
Мы провели анализ своевременности установления диагноза у вновь выявленных больных с адреногенитальном синдромом, находившихся на лечении в Республиканском детском эндокринологическом центре (на базе 2-й ГДКБ Минска) в 2006–2009 гг. Оказалось, что сроки диагностики сольтеряющей формы адреногенитального синдрома для девочек составили 8,6±0,4 дня, для мальчиков — 26,7±17,9 дня. Во всех случаях диагноз ставился на фоне развивающегося сольтеряющего криза при уровнях калия 7,8±1,6 ммоль/л, натрия — 128,6±0,9 ммоль/л и декомпенсированном метаболическом ацидозе.
Вирильная форма заболевания определялась поздно: у девочек в возрасте 1,2±0,1 года, у мальчиков — 6,0±0,1 года. У 50–60% мальчиков она вообще не выявляется. Так, в группе детей с вирильным вариантом заболевания девочки составили 82%, мальчики — 18% (согласно аутосомно-рецессивному типу наследования АГС, их число должно быть почти равным). Результаты нашего исследования совпадают с литературными данными. Неонатальный скрининг на адреногенитальный синдром целесообразно внедрить в Беларуси.
Основной метод терапии классической формы адреногенитального синдрома основан на применении глюкокортикоидов, подавляющих гиперсекрецию кортикотропин-рилизинг гормона и АКТГ и нормализующих выработку андрогенов надпочечниками. Цель лечения — в замещении дефицита стероидов, секреция которых снижена из-за ферментативного дефекта, в оптимизации роста пациентов, обеспечении нормального полового созревания и потенциальной фертильности.
Препаратом выбора при терапии адреногенитального синдрома у детей с открытыми зонами роста является гидрокортизон. Обычно у новорожденных лечение начинают с массивной стартовой дозы 50–100 мг внутривенно (до 400 мг/м2 в сутки), что отражает высокую скорость секреции кортизола в этом возрасте. Но следует избегать длительного применения у ребенка ввиду негативного влияния на процессы роста. При переводе больного на таблетированный препарат суточная доза снижается до 15–20 мг/м2. Адекватная заместительная терапия глюкокортикоидами нормализует гормональные показатели за несколько недель.
Ребенку старше 2 лет гидрокортизон назначается из расчета 10–15 мг/м2 в сутки. Препарат дается 3 раза в день в равных частях или в пропорции 0,25:0,25:0,5. У пациентов с зонами роста, близкими к закрытию, применяют глюкокортикоиды длительного действия с более выраженным АКТГ-подавляющим эффектом (преднизолон или дексаметазон). Доза преднизолона составляет 2–4 мг/м2 в сутки, при этом 1/3 ее назначается утром, 2/3 — перед сном. Суточное количество дексаметазона (0,25–0,35 мг/м2) дается однократно в вечерние часы. Ранее популярный при заместительной терапии АГС кортизона ацетат проявляет активность только после конверсии в активный кортизол под действием печеночного фермента 11ß-гидроксистероиддегидрогеназы. В некоторых случаях этот механизм может иметь функциональный или частичный дефект, особенно в первые месяцы постнатальной жизни ребенка, или большую индивидуальную вариабельность. В связи с этим применение кортизона ацетата при лечении больных с дефицитом 21-гидроксилазы нежелательно.
Коррекция дозы глюкокортикоидов проводится под контролем сывороточных концентраций 17-OHП. У мальчиков в периоде допубертата и женщин дополнительно исследуют уровни тестостерона. Гормональные показатели определяют в соответствии с режимом назначения медикаментов, предпочтительно в 8 часов утра с учетом физиологического пика секреции АКТГ или на самом низком уровне кортизона в крови — непосредственно перед вечерним приемом глюкокортикоидов. Наблюдение больных с дефицитом 21-гидроксилазы должно включать обязательный ежегодный анализ динамики роста и контроль костного возраста.
Высокие дозы глюкокортикоидов, особенно в раннем возрасте, могут быть причиной снижения роста ребенка, развития ожирения, повышения артериального давления или других проявлений синдрома Кушинга.
Всем пациентам с классической сольтеряющей формой АГС после установления диагноза наряду с глюкокортикоидами назначается флюдрокортизона ацетат. Суточная доза для детей первого года жизни — 0,05–0,3 мг в зависимости от уровня электролитов. У старших ребятишек потребность в минералокортикоидах снижается до 0,05–0,15 мг в сутки с последующей отменой. Необходимость в препарате у ребенка определяется на основании оценки уровней ренина плазмы и артериального давления.
Из клинических наблюдений известно, что в пубертатный период у детей с АГС не удается достичь компенсации заболевания, несмотря на рекомендуемую оптимальную дозу глюкокортикоидных препаратов. Традиционно этот факт объяснялся психосоциальными причинами, характерными для подросткового возраста: отрицанием авторитетов, увеличением степени собственной свободы и независимости, снижением контроля со стороны родителей за приемом лекарств, нарушением режима введения и дозы глюкокортикоидов вплоть до полного прекращения заместительной терапии.
Но существенная причина — в изменении фармакокинетики кортизола в периоде пубертата: увеличение клиренса и укорочение периода жизни под влиянием повышения ростовых факторов, половых гормонов (эстрадиола). Изменение распределения кортизола в организме ведет к гипокортизолемии при неизменной дозе глюкокортикоидов.
У больных с адреногенитальным синдромом в периоде пубертата есть риск передозировки глюкокортикоидов из-за развития выраженной инсулинорезистентности смешанного генеза (физиологической и на фоне избыточной массы тела), что диктует увеличение кратности приема гидрокортизона при неизменной суточной дозе препарата.
Случай из практики
Девочка Е. от 2-й доношенной беременности, вес при рождении 3 370 г, рост 53 см. Выписана на 5-е сутки из роддома в удовлетворительном состоянии. Со слов матери, дома буквально на следующий день появились срыгивания после каждого кормления, вялое сосание, учащенный стул. Обратились в приемное отделение 2-й ГДКБ Минска, когда ребенку было 15 дней. При осмотре: состояние тяжелое, обусловленное дегидратацией; кожные покровы и слизистые сухие; гиперпигментация сосков. Потеря массы тела с момента рождения — 670 г. Наружные гениталии имели бисексуальное строение: отмечалось сращение больших половых губ, гипертрофия клитора с формированием головки, синус урогениталис. При лабораторном обследовании выявлены гиперкалиемия (калий — 7,06 ммоль/л), гипонатриемия (122,9 ммоль/л), гипохлоремия (91,8 ммоль/л), гипогликемия (2,8 ммоль/л). Показатели кислотно-основного состояния в норме. Уровень 17-ОНП повышен — 18 нг/мл (норма до 3,0). На основании данных обследования поставлен диагноз: адреногенитальный синдром, сольтеряющая форма, состояние клинико-метаболической декомпенсации, гипотрофия 2-й степени. Назначена заместительная терапия препаратами глюкокортикоидного ряда и минералокортикоидами.
Анжелика Солнцева, доцент 1-й кафедры детских болезней БГМУ, главный внештатный детский эндокринолог Минздрава
Медицинский вестник, 10 февраля 2011
Надпочечниковая вирилизация
При надпочечниковой вирилизации вирильный синдром обусловлен избыточной секрецией надпочечниковых андрогенов. Диагноз устанавливается клинически и подтверждается повышенным уровнем андрогенов; выяснение причины может требовать визуализации надпочечников. Лечение зависит от этиологии заболевания.
Причины адренального вирилизма следующие:
Андроген-секретирующие опухоли надпочечников
Злокачественные опухоли надпочечников могут производить избыток андрогенов, кортизола или минералокортикоидов (или всех трех гормонов одновременно). Если происходит избыточная секреция кортизола , это приводит к развитию синдрома Кушинга Синдром Кушинга Синдром Кушинга – это сочетание клинических симптомов, вызванных хроническим повышением уровня кортизола или родственных ему кортикостероидов в крови. Болезнь Кушинга – это синдром Кушинга. Прочитайте дополнительные сведенияГиперплазия надпочечников – обычно врожденная патология; постпубертатная вирилизующая гиперплазия надпочечников представляет собой вариант врожденной гиперплазии надпочечников Обзор врожденной дисфункции коры надпочечников (Overview of Congenital Adrenal Hyperplasia) Врожденная гиперплазия надпочечников представляет собой группу наследственных заболеваний, каждое из которых характеризуется недостаточным синтезом кортизола, альдостерона или обоих гормонов. Прочитайте дополнительные сведения . В обоих этих случаях нарушено гидроксилирование предшественников кортизола, которые накапливаются и вступают в реакции образования андрогенов. При постпубертатной вирилизующей гиперплазии надпочечников такое гидроксилирование нарушается лишь частично, и клинические признаки заболевания могут появиться лишь в зрелом возрасте.
Симптомы и признаки надпочечниковой вирилизации
Проявления зависят от пола больного и возраста начала заболевания; у женщин они выражены гораздо отчетливее, чем у мужчин.
В препубертатном возрасте отмечается ускоренный рост. В отсутствие лечения происходит преждевременное закрытие эпифизарных зон роста и больные остаются низкорослыми. У мальчиков наблюдается преждевременное половое развитие.
Для взрослых женщин характерны аменорея, атрофия матки, гипертрофия клитора, уменьшение размеров молочных желез, угревая сыпь, гирсутизм, низкий голос, облысение, повышенное либидо и сильно развитая мускулатура.
У взрослых мужчин избыток андрогенов надпочечников может подавлять функцию половых желез и вызывать бесплодие. Эктопическая надпочечниковая ткань в семенниках может увеличиваться, имитируя наличие опухоли.
Диагностика адренального вирилизма
Определение уровня тестостерон
Определение уровней других надпочечниковых андрогенов (дегидроэпиандростерона [ДГЭА] и его сульфата [ДГЭАС], андростендиона)
Супрессивный тест с дексаметазоном
Иногда тест на стимуляцию адренокортикотропным гормоном (АКТГ)
Адренальный вирилизм может быть заподозрен на основании клинических данных, хотя легкий гирсутизм и вирилизация с гипоменореей и повышением уровня тестостерона в плазме встречается и при синдроме поликистоза яичников Cиндром поликистозных яичников (СПЯ) Синдром поликистозных яичников – клинический синдром, характеризующийся умеренным ожирением, нерегулярными менструациями или аменореей и признаками избытка андрогенов (например, гирсутизм, угри). Прочитайте дополнительные сведения (синдром Штейна-Левенталя). Диагноз подтверждает повышенный уровень надпочечниковых андрогенов.
При гиперплазии надпочечников повышено содержание дегидроэпиандростерона (ДГЭА) и его сульфата (ДГЭАС) в моче; нередко возрастает и экскреция прегнантриола (метаболита 17-гидроксипрогестерона), тогда как уровень свободного кортизола в моче нормален или снижен. Содержание ДГЭА, ДГЭАС, 17-гидроксипрогестерона, тестостерона и андростендиона в плазме обычно повышено. Уровень 17-гидроксипрогестерона > 30 нмоль/л (1000 нг/дл) через 30 минут после в/м введения 0,25 мг косинтропина (синтетического АКТГ) с высокой вероятностью свидетельствует о наиболее распространенной форме гиперплазии надпочечников.
Подавление избыточной продукции андрогенов при приеме 0,5 мг перорально дексаметазона каждые 6 часов в течение 48 часов позволяет исключить вирилизующую опухоль надпочечников. Если же избыточная экскреция андрогенов сохраняется, то в поисках опухоли проводят КТ или МРТ надпочечников и УЗИ яичников.
Лечение вирилизации надпочечников
При гиперплазии – прием глюкокортикоидов перорально
При гиперплазии надпочечников назначают глюкокортикоиды, как правило, пероральный гидрокортизон 10 мг при пробуждении, 5 мг в середине дня и 5 мг ближе к вечеру. Как альтернативный вариант, возможен пероральный прием дексаметазона по 0,5–1,0 мг перед сном, но даже столь небольшие дозы могут вызывать признаки синдрома Кушинга, поэтому данная форма глюкокортикоида, как правило, не рекомендуется. Прием дексаметазона перед сном лучше всего подходит для подавления секреции АКТГ, но может вызвать бессонницу. Вместо этого можно принимать кортизона ацетат по 25 мг перорально 1 раз в день или преднизон по 5 или периодически до 10 мг перорально 1 раз в день. Хотя лечение устраняет большинство симптомов и признаков вирилизации, гирсутизм и облысение исчезают медленно, голос может оставаться низким, а бесплодие – сохраняться.
При опухолях производят адреналэктомию. Если опухоль секретирует кортизол , то перед и после операции следует назначать гидрокортизон, поскольку нормальная ткань коры надпочечников в таких случаях атрофирована, а ее функция подавлена.
Основные положения
В основе адреногенитального синдрома лежит андрогенсекретирующая опухоль надпочечника или гиперплазия надпочечников.
Вирилизация особенно заметна у женщин; у мужчин может иметь место бесплодие из-за угнетения функции половых желез.
Содержание дегидроэпиандростерона (ДГЭА) и его сульфата (ДГЭАС) в моче и плазме, а часто и тестостерона в плазме повышено.
Для диагностики можно использовать супрессивный тест с дексаметазоном и/или стимуляционный тест с адренокортикотропным гормоном (АКТГ).
В лечении гиперплазии применяют кортикостероидную терапию; опухоли требуют адреналэктомии.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Заболевания надпочечников
Лечение заболеваний надпочечников Надпочечники – эндокринные железы, которые располагаются на верхнем полюсе обеих почек, состоят из коркового и мозгового слоев и отвечают за выработку ряда стероидных гормонов и катехоламинов. В корковом слое надпочечников синтезируются следующие группы гормонов:
• Глюкокортикоиды (основной представитель – кортизол), оказывающие на организм антистрессовое, противошоковое, имунорегулирующее, противовоспалительное, противоаллергическое действие и влияющие на метаболизм веществ и выработку других гормонов;
• Минералокортикоиды (в основном альдостерон), конечное действие которых заключается в увеличении объема циркулирующей крови и повышении уровня артериального давления;
• Андрогены – только после преобразования в тестостерон и дигидротестостерон проявляют свое основное физиологическое действие, характерное для половых гормонов.
Мозговой слой надпочечников отвечает за синтез:
• Катехоламины (адреналин и норадреналин), влияющие на работу сердца, артериальное давление, также расширяют просвет бронхов, увеличивают липолиз, активируют гликогенолиз и выработку ренина.
Болезни надпочечников
Классификация заболеваний надпочечников основана на характере нарушений функционального состояния органа, которая может выражаться либо в увеличении секреции гормонов, либо в снижении, либо в дисфункции.
• Гипофункция может быть первичной (при болезни Аддисона) или вторичной (гипоталамо-гипофизарная недостаточность);
• Гиперфункция проявляется различными клиническими синдромами. При гиперсекреции андрогенов развивается надпочечниковая вирилизация, глюкокортикоидов – синдром Кушинга, альдостерона – гиперальдостеронизму. Избыток секреции катехоламинов характерен для феохромоцитомы;
• Дисфункция коры надпочечников (синдром Прадера и др.)
Болезнь Аддисона
Лечение заболеваний надпочечников в Кривом Роге Это хроническая адренокортикальная недостаточность – постепенно развивающаяся, прогрессирующая функциональная недостаточность коры надпочечников. В большинстве случаев причина неизвестна. Связывают с идиопатической атрофией коры надпочечников, вызванной аутоиммунными процессами. Частота заболевания не превышает 5 человек на 100 тыс. населения в год.
Клинические признаки болезни Аддисона
Проявляется различными симптомами, включая слабость, утомляемость, головокружение, обморочные состояния, тошноту, рвоту, потерю аппетита, пониженное артериальное давление, потемнение кожных покровов (гиперпигментация). Состояние может ухудшаться до надпочечникового криза с сердечнососудистым коллапсом. На более поздних стадиях прослеживается потеря веса, обезвоживание и гипотензия.
Диагностика болезни Аддисона
Основной диагностический критерий при постановке диагноза болезнь Аддисона является обнаружение повышенного уровня АКТГ (адренокортикотропный гормон) и низкого уровня кортизола в плазме крови. Для установления заболевания и его подтверждения, необходимо обратиться к эндокринологу, который соберет анамнез, проведет осмотр и назначит лабораторные исследования!
Лечение заболевания
Как правило, лечение болезни Аддисона сводится к применению гидрокортизона. В некоторых случаях, назначаются и другие гормоны. Также, в качестве препарата для замещения гормонов надпочечников, врач может назначить флудрокортизон и другие средства. Обязательно проводится коррекция дозировок. Врач рекомендует специальную диету для скорейшего восстановления организма!
Вторичная надпочечниковая недостаточность
Гипофункция надпочечников, которая вызвана недостатком продукции АКТГ и может наблюдаться при пангипопитуитаризме, при длительном приеме или после прекращении приема глюкокортикоидов. Сниженная выработка АКТТ также может обусловлена нарушением гипоталамо-гипофизарной системы регуляции синтеза гормонов.
• Клинические признаки практически идентичны симптомам болезни Аддисона;
• Диагностика. Для определения вида надпочечниковой недостаточности (первичная или вторичная) проводят специальные тесты. Золотым стандартом для проверки состояния гипоталамо-гипофизарно-надпочечникой системы считается инсулиновый стрессовый тест, провоцирующий снижение уровня сахара в крови и повышения уровня кортизола;
• Лечение вторичной надпочечниковой недостаточности. В качестве терапии чаще используют гидрокортизон!
Надпочечниковая вирилизация
Диагностика заболеваний надпочечников Клинический синдром, при котором наблюдается маскулинизация, связанная с повышенной продукцией андрогенов сетчатой зоной коры надпочечников. Причиной развития данного синдрома может быть опухоль или гиперплазия надпочечников.
• Клиническая симптоматика зависит от пола и возраста пациента в момент дебюта заболевания. Более заметны симптомы вирилизации у женщин. Характерно гирсутизм (чрезмерное оволосение по мужскому типу), облысение, акне, огрубение голоса. Девочки страдают аменореей, атрофией матки, увеличением в размерах клитора, уменьшением груди, появлением вторичных мужских половых признаков. У мальчиков отмечается раннее половое созревание, у мужчин – подавление функции половых желез и потеря репродуктивной способности;
• Диагностика. Характерные клинические данные дают основания заподозрить надпочечниковую вирилизацию. Диагноз подтверждается по выявлению высокого уровня андрогенов надпочечников;
• Лечение надпочечниковой вирилизации зависит от причины заболевания. При опухолях проводят оперативное вмешательство, при гиперплазии назначают гормональную терапию.
Синдром Кушинга: лечение в Кривом Роге
Комплекс клинических симптомов, обусловленный длительно высоким уровнем глюкокортикоидов в плазме крови. Появление избытка глюкокортикоидов в организме может быть связано как с повышенной эндогенной его продукцией, так и с продолжительным экзогенным поступлением в виде лекарственных препаратов.
• Клиническая картина: лунообразное лицо, кушингоидный тип ожирения (отложение жира на животе, груди, шее, лице), атрофия мышц, тонкие конечности, синюшные стрии на животе, гипертензия, кардиомиопатия, нефролитиаз, остеопороз;
• Диагностика синдрома Кушинга базируется на клинических симптомах, данных анамнеза (длительный прием глюкокортикоидов) или определении высокого уровня кортизола в анализе крови;
• Лечение синдрома Кушинга комплексное, и зависит от причины заболевания. Чаще назначается медикаментозная терапия, направленная на блокировку повышенной выработки кортикостероидов и АКТГ!
Гиперальдостеронизм: диагностика и лечение
Феохромоцитома: лечение в Кривом Роге
Феохромоцитома представляет собой опухоль клеток мозгового вещества надпочечников, приводящая к избыточному продуцированию катехоламинов.
• Клиника и диагностика. Высокий уровень катехоламинов в крови вызывает постоянную или приступообразную гипертензию. Подтвердить диагноз помогает измерение катехоламинов и их метаболитов в крови или моче, а также проведение КТ или МРТ для обнаружения опухоли;
• Лечение феохромоцитомы. По возможности желательно искоренить причину заболевания – удалить опухоль. Контроль АД проводится за счет назначения альфа-блокаторов или в комбинации с бета-блокаторами.
УЗИ почек и надпочечников Наши специалисты используют в своем арсенале современное ультразвуковое и лабораторное оборудование для проведения диагностики заболеваний надпочечников в Кривом Роге, позволяющее определить их на ранних стадииях. Консультация эндокринолога и других профильных врачей, экспертный диагностический комплекс и назначение индивидуальной терапии патологий надпочечников на Филатова, Клиника Медитон!
Надпочечниковая вирилизация
Преднизолон 8,75 мг/сут; Флудрокортизон 50 мкг/сут
Коплексное уродинамическое исследование
Мочеиспускание от 5 до 11 раз в день, порциями по 150—200 мл. Ежедневно отмечает императивные позывы на мочеиспускание, подтекание мочи 1 раз в неделю, в том числе ночью, порциями 20—30 мл. При исследовании нормосенсорный, нормоконтактный мочевой пузырь с увеличением уретрального давления до 114 мм вод. ст., увеличением функциональной длины уретры до 10 см и необструктивным типом мочеиспускания
Мочеиспускание от 3 до 4 раз в день, порциями по 200—350 мл без императивных позывов и недержания мочи. При исследовании гиперсенсорный, нормоконтактный мочевой пузырь с увеличением уретрального давления до 320 мм вод. ст., увеличением функциональной длины уретры до 11 см и необструктивным типом мочеиспускания
Пациентка 2. Диагноз ВДКН заподозрен при рождении в связи с вирилизацией НПО по Prader IV степени. Однако дополнительное обследование не проведено. В течение 1-го месяца в связи с обильными срыгиваниями с 15-го дня жизни потеря массы тела составила 800 г. Девочка госпитализирована в стационар в возрасте 1 мес в тяжелом состоянии с признаками эксикоза. При обследовании выявлен нормальный женский кариотип 46,XX, определены повышенные значения 17-ОНП — 500 нмоль/л, ренина — 238 мЕд/л, гиперкалиемия и гипонатриемия. На основании результатов проведенного обследования установлен диагноз «ВДКН, дефицит 21-гидроксилазы, сольтеряющая форма» и назначена заместительная гормональная терапия минерало- и глюкокортикоидами, с положительным эффектом. Диагноз подтвержден молекулярно-генетическим исследованием: выявлена гомозиготная мутация 12spl в гене CYP 21, характерная для сольтеряющей формы заболевания. В возрасте 5 лет проведен первый этап феминизирующей пластики (клиторопластика, синусотомия). До 13 лет жизни отмечалось относительно стабильное течение заболевания. Однако в 13 лет девочка самостоятельно прекратила прием препаратов на 4 мес. В течение следующих 1,5 лет, несмотря на постоянную коррекцию проводимой терапии, не удалось добиться компенсации заболевания. Показатели гормонов в периоды декомпенсации представлены в табл. 1. На момент поступления в стационар в возрасте 16 лет: рост 165,4 см (SDS роста 0,47), масса тела 58 кг, телосложение гиперстеническое, маскулинное, голос низкий, хорошо выраженная мышечная система, половое развитие по Таннер (В3, P 4), менструации отсутствуют. Данные статуса НПО, лабораторных исследований и получаемая терапия при поступлении в стационар указаны в табл. 2.
С целью определения объема и тактики оперативного лечения в обоих случаях проведено магнитно-резонансное томографическое (МРТ) исследование органов малого таза без контрастирования в режимах Т1, Т2, DW, выполненное на оборудовании Optima MR450w №415449 («GE Healthcare», США). В области уретры, проксимальнее уретровагинального конфлюенса выявлена ткань ПЖ, имеющая небольшой объем и отсутствие зональной дифференцировки. На МР-изображениях ткань ПЖ располагается типично для мужской ПЖ в парауретральной области, за лобковым симфизом, книзу от мочевого пузыря, эллипсоидной формы, представлена преимущественно железистой тканью периферической зоны, однородной структуры с гиперинтенсивным гомогенным сигналом в режиме Т2-ВИ и изоинтенсивным сигналом в режиме Т1-ВИ (рис. 1, 2). Комплексное уродинамическое исследование (КУДИ) не выявило признаков инфравезикальной обструкции (см. табл. 2). При цистоуретровагиноскопии, проведенной непосредственно перед операцией, выявлены высокий уретровагинальный конфлюенс и признаки гранулярного цистита в области треугольника Льетто у обеих девочек. Особенностью патологического строения мочеполовых путей у пациентки 2 явилось резкое сужение входа во влагалище. Последний открывался отверстием диаметром 0,3 см, окруженным складкой слизистой (гименальным кольцом) над уровнем тазовой диафрагмы. Показания к проведению второго этапа феминизации у пациентки 1 обоснованы в связи с наступлением первой менструации и достаточной эстрогенизацией гениталий [3]. Пациентка 2 не достигла менархе. С учетом эндоскопической находки в виде резкого сужения входа во влагалище и риска развития клиники гематокольпос и гематометра при наступлении менархе в будущем вследствие нарушения оттока менструальных выделений принято решение о проведении второго этапа феминизации. Пациенткам проведена разобщающая мочевые и половые пути интроитопластика. Выполнена полная мобилизация общего мочеполового канала (ОМК) с формированием передней стенки искусственного входа во влагалище (ИВВ) слизистым лоскутом в модификации Passorini—Glasel. Задняя стенка ИВВ сформирована с помощью омега-образного кожного лоскута промежности. Неоуретра и неомеатус сформированы из участка ОМК. Пациентки выписаны из стационара в удовлетворительном состоянии на 9-е и 11-е сутки после операции. Рекомендовано следующее: гормональная терапия, получаемая пациентками на момент поступления (см. табл. 2); ежеквартальный контроль уровней 17-ОНП, тестостерона, АКТГ и ренина плазмы крови для оценки компенсации глюкокортикоидной и минералокортикоидной недостаточности; наблюдение гинеколога; контрольная МРТ малого таза; уродинамическое исследование через 6—12 мес для оценки результата операции и динамики роста ПЖ; ежегодный контроль уровня простатического специфического антигена (ПСА) и повторная МРТ в случае его увеличения.
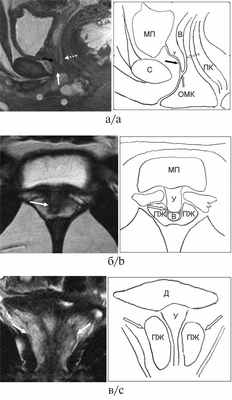
Рис. 1. Серия МРТ малого таза в режиме Т2-ВИ со скицами пациентки 1, 17 лет, с диагнозом «ВДКН, сольтеряющая форма».
а — сагиттальная плоскость; б — аксиальная плоскость; в — фронтальная плоскость. На изображениях визуализируются уретра (черная стрелка), влагалище (белая пунктирная стрелка) с наличием в просвете небольшого количества жидкости. Уретровагинальный конфлюенс (белая стрелка) определяется на уровне средней трети симфиза. Круглыми МРТ метками на рис. а обозначены наружное отверстие общего мочеполового канала, расположенное ближе к симфизу, и проекция входа во влагалище, расположенного ближе к прямой кишке. Визуализируется ткань предстательной железы (головки черных стрелок), эллипсоидной формы, без четкой зональной дифференцировки, размерами 34×15×32 мм (объем 8,4 см 3 ) без видимых очаговых изменений в структуре. МП — мочевой пузырь, С — симфиз, У — уретра, В — влагалище, ОМК — общий мочеполовой канал, ПК — прямая кишка, ПЖ — предстательная железа, Д — детрузор мочевого пузыря.
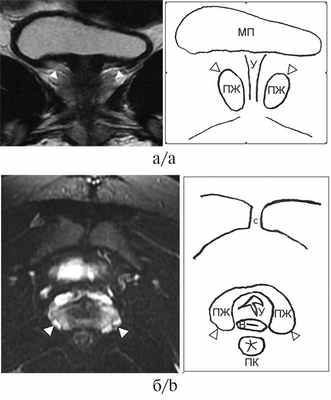
Рис. 2. Серия МРТ малого таза со скицами пациентки 2, 16 лет, с диагнозом «ВДКН, сольтеряющая форма».
а — корональная плоскость в режиме Т2-ВИ; б — аксиальная плоскость в режиме Т2-ВИ с подавлением сигнала от жировой ткани. Визуализируется ткань предстательной железы (белые головки стрелок), эллипсоидной формы, без четкой зональной дифференцировки, размерами 30×14×21 мм (объем 4,6 см 3 ). Структура предстательной железы представлена преимущественно железистой тканью периферической зоны, однородной структуры с гиперинтенсивным гомогенным сигналом без видимых очаговых изменений. МП — мочевой пузырь, С — симфиз, У — уретра, В — влагалище, ПК — прямая кишка, ПЖ — предстательная железа.
R. de Graaf, голландский физиолог и гистолог, впервые описал структуру и функцию парауретрального органа и присвоил ему термин «женская простата» в 1672 г. [4]. Спустя примерно 200 лет в 1880 г. американский гинеколог A. Skene описал женскую простату, состоящую из двух основных парауретральных протоков, открывающихся с обеих сторон уретрального отверстия [5]. Многочисленные исследования этого органа женской мочеполовой системы проведены в последующем. S. Tepper и соавт. в 1984 г. доказали с помощью гистохимического исследования гомологичную природу женских парауретральных желез (ПУЖ) и ПЖ [6]. M. Zaviacic и R. Ablin в 2000 г. в обзорной статье представили дополнительные доказательства идентичности ПУЖ и мужской простаты на клеточном и ферментативном уровнях [7]. Авторы, опираясь на исторические факты и собственные данные исследований, предложили утвердить термин «женская простата» для использования в литературе. Эмбриологические исследования, проведенные за длительный период, начиная с середины XX века, также подтвердили сходство ПЖ и ПУЖ. Эмбриологическая закладка желез начинается на 10—11-й неделе внутриутробного периода. На вентральной поверхности урогенитального синуса, который присутствует у эмбрионов обоих полов, появляются несколько рядов солидных выростов в окружающую мезенхиму. Под воздействием андрогенов, вырабатываемых яичками мужского плода, постепенно количество выростов увеличивается. Достигнув определенной глубины, выросты ветвятся и канализируются, приобретают железистый и мышечный компонент, создавая группы, соответствующие долям ПЖ. У женского плода ПУЖ приобретают типичное для них строение в виде нескольких протоков с железистой тканью [8, 9].
Закладка коры надпочечников происходит на 11-й неделе. В течение этого периода у женского плода с ВДКН избыток надпочечниковых андрогенов стимулирует вирилизацию НПО и, что подтверждено опытами на животных, рост ПУЖ [8]. У некоторых девочек с ВДКН при МРТ исследовании визуализируют ПЖ [10]. Как трактовать эти находки? Как ПЖ или как гипертрофированные ПУЖ? Ответ на этот вопрос остается неясным.
Если эмбриогенез и рост ПЖ/ПУЖ очевиден, почему она выявляется не у всех девочек с ВДКН? Этот вопрос также остается без ответа. Однако гипотеза об андрогензависимости этого органа у девочек с ВДКН упоминается в двух проспективных исследованиях. M. da C. Paulino и соавт. в 2009 г. в группе из 36 пациенток с ВДКН на фоне искусственно созданной гиперандрогении выявили ткань ПЖ у 15,6% [10]. Напротив, исследование P. Doherty и соавт., нацеленное на поиск ткани ПЖ, не выявило ее [11]. Однако малая выборка (всего 11 пациенток) и их однородный контингент (только женщины кавказского происхождения) делают данное исследование малоинформативным.
Благодаря своей способности к многоплоскостной визуализации и превосходному контрасту мягких тканей МРТ является наиболее чувствительным методом обнаружения доброкачественной и злокачественной патологии уретры. Нормально развитая ткань ПУЖ не визуализируется на МРТ [12]. Увеличение ее размера >5 мм может быть зафиксировано и трактоваться как ПЖ в связи с известной формой, структурой и положением, описанными у пациентов мужского пола. В наших случаях у пациенток с ВДКН ПЖ на МР изображениях имела характерную форму, локализацию и структуру объемом 8,4 и 4,6 см 3 . При этом выявленная железа не влияла на опорожнение мочевого пузыря. Данные КУДИ не выявили нарушения уродинамики нижних мочевых путей, что свидетельствовало об отсутствии патологического роста ПЖ.
В доступных источниках литературы мы не обнаружили рекомендации по ведению пациенток с ВДКН и ПЖ. Учитывая, что в литературе описаны 4 случая аденокарциномы простаты в возрасте от 60 до 88 лет [13—16] и 1 случай доброкачественной гиперплазии ПЖ в возрасте 60 лет [17], мы считаем необходимым наблюдение данной категории пациенток с целью своевременной коррекции терапии.
Заключение
Наличие ПЖ у девочек с кариотипом 46,XX и ВДКН является редким вариантом патологического развития мочеполовых путей. С учетом риска возникновения аденокарциномы и доброкачественной гиперплазии ПЖ таким пациенткам вместе с контролем компенсации заболевания следует проводить ежегодный контроль уровня простатического специфического антигена и в случае его увеличения — МРТ органов малого таза. Нет четких данных, которые позволили бы однозначно трактовать находки на МРТ у данного контингента пациенток — ПЖ это или гипертрофированные ПУЖ. Нет достаточных доказательств причины гипертрофии ПУЖ. Данные литературы указывают на гиперандрогению как причину увеличения ПУЖ, но есть и противоположные точки зрения. Непонятно, почему усиленная выработка андрогенов, обусловливающая вирилизацию наружных половых органов, характерная для врожденной дисфункции коры надпочечников, не приводит к увеличению ПУЖ у всех больных этой группы. Не разработаны рекомендации по ведению данных пациенток. Поскольку четко не разработаны аспекты, связанные с причинами развития ПЖ у девочек с ВДКН, частотой ее выявления и рекомендациями по ведению таких пациентов, данный вопрос требует дальнейшего изучения.
Согласие пациента
Медицинские данные опубликованы с письменного согласия пациентов.
Участие авторов:
Концепция и дизайн исследования — Андреева Е.Н.
Сбор и обработка материала — Аникиев А.В., Калинченко Н.Ю., Бабаева Д.М., Владимирова В.П.
Написание текста — Аникиев А.В., Калинченко Н.Ю., Бабаева Д.М., Владимирова В.П.
Адреногенитальный синдром
Адреногенитальный синдром (АГС) также известен как врожденная дисфункция коры надпочечников (ВДКН) или врожденная гиперплазия коры надпочечников (ВГКН). Синдром объединяет заболевания с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов метаболического пути синтеза стероидов.
Заболевание вызвано наследственным дефицитом ферментов, которые локализованы в коре надпочечников. Гормоны коры надпочечников включают минералокортикоиды (альдостерон), глюкокортикоиды (кортизол) и половые стероиды (тестостерон и эстроген). Синдром возникает, когда дефицит фермента приводит к снижению надпочечникового синтеза глюкокортикоидов. Происходит снижение ингибирующего влияния гормонов коры надпочечников на гипофиз, поэтому синтез и секреция адренокортикотропного гормона (АКТГ) увеличивается по механизму отрицательной обратной связи. АКТГ стимулирует увеличение размеров надпочечников и выработку ими промежуточных субстратов. Включаясь в другие пути синтеза, метаболиты вызывают повышение уровней других гормонов коры надпочечников — минералокортикоидов или андрогенов. Измененные уровни минералокортикоидов и половых гормонов приводят к аномалиям электролитного соотношения, проблемам с дифференциацией пола и другим признакам, и симптомам, в зависимости от дефицита фермента и его степени.
Частота встречаемости заболевания от 1:5000 до 1:67000.
Характеристика заболевания
ВГКН является одним из самых распространенных наследственных моногенных заболеваний, одновременно представляет собой вариант хронической первичной надпочечниковой недостаточности и группу патологии полового развития, а также половой дифференцировки. АГС включен в программу «Национальные приоритетные проекты» и введен в неонатальный скрининг. АГС в стертой (неклассической) форме является одной из причин нарушения репродуктивного здоровья (бесплодие, невынашивание беременности).
Дефект 21-гидроксилазы: классификация и клинические проявления
Ген CYP21, кодирующий фермент 21-гидроксилазу, локализован на коротком плече 6-й хромосомы. Описано более пятидесяти мутаций этого гена, приводящих к синтезу фермента со степенью активности от 0 до 60 %.
Патогенетической сущностью синдрома является нарушение процесса перехода 17-гидроксипрогестерона и 11-дезоксикортикостерона в 11-дезоксикортизол (рис 1). Развивается гормональная дисфункция, которая сводится к снижению синтеза кортизола альдостерона и избыточному накоплению предшественников кортизола. Избыток прегненолона, прогестерона, 17-гидроксипрогестерона конвертируется в надпочечениковые андрогены.
Рис.1. Недостаточность 21-гидроксилазы. Штриховкой выделены стероиды, синтез которых заблокирован.
Так происходит угнетение выработки одних кортикостероидов при одновременном увеличении выработки других, вследствие дефицита того или иного фермента, обеспечивающего один из этапов стероидогенеза.
- дефицит 21-гидроксилазы с сольтеряющим синдромом;
- простая вирильная форма (неполный дефицит 21-гидроксилазы);
- неклассическая форма (постпубертатная).
Симптомы недостаточности 21-гидроксилазы
При классической вирильной форме адреногенитального синдрома, наружные половые органы у девочек сформированы по гетеросексуальному типу — гипертрофирован клитор, большие половые губы напоминают мошонку, вагина и уретра представлены урогенитальным синусом. У новорождённых мальчиков явных нарушений выявить не удаётся. С 2–4 лет у детей обоего пола появляются другие симптомы адреногенитального синдрома, то есть андрогенизации: формируется подмышечное и лобковое оволосение, развивается скелетная мускулатура, грубеет голос, маскулинизируется фигура, появляются юношеские угри на лице и туловище. В пубертатном периоде у девочек не растут молочные железы, не появляются менструации. При этом ускоряется дифференцировка скелета, и зоны роста закрываются преждевременно, это обусловливает низкорослость.
При сольтеряющей форме 21-гидроксилазной недостаточности, помимо вышеописанных симптомов, у детей с первых дней жизни отмечают признаки надпочечниковой недостаточности. Появляются вначале срыгивания, затем рвоты, возможен жидкий стул. Ребёнок быстро теряет массу тела, развиваются симптомы дегидратации, нарушения микроциркуляции, снижается артериальное давление, начинается тахикардия, возможна остановка сердца вследствие гиперкалиемии.
Неклассическая форма адреногенитального синдрома характеризуется ранним появлением вторичного оволосения, ускорением роста и дифференцировки скелета. У девочек пубертатного возраста возможны умеренные признаки гирсутизма, нарушения менструального цикла (первый цикл может запаздывать или начаться раньше времени). В позднем возрасте при небольших дефектах гена на адреногенитальный синдром может указывать наличие бесплодия в анамнезе. Менструальный цикл может быть нерегулярным и иметь тенденцию к задержке. Невынашивание беременности при адреногенитальном синдроме встречается у четверти женщин с дефектным геном.
- женщинам с диагнозом «замершая эмбриональная беременность»;
- женщинам с привычным невынашиванием беременности;
- женщинам с диагнозом СПКЯ неустановленной этиологии;
- девушкам пубертатного возраста с проявлениями неклассической формы ВДКН: олигоменореей, гирсутизмом, акне и интерсексуальным типом телосложения;
- девочкам младшего возраста с вирилизацией наружных гениталий для дифференциальной диагностики ВДКН с идиопатической врожденной вирилизацией наружных гениталий;
- детям младшего возраста (2–4 года) с признаками преждевременного полового созревания по мужскому типу для дифференциальной диагностики вирильной формы ВДКН с надпочечниковой недостаточностью, гермафродитизмом другого генеза, различными вариантами преждевременного полового созревания и андрогенпродуцирующей опухолью надпочечников.
40.131 Анализ полиморфизмов гена стероид-21-гидролаза (CYP21,9 точек) (адреногенитальный синдром)
Читайте также: