Наследственные аритмии. Лечение сердечных нарушений синдрома Марфана
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Синдром Марфана: причины, диагностика, лечение
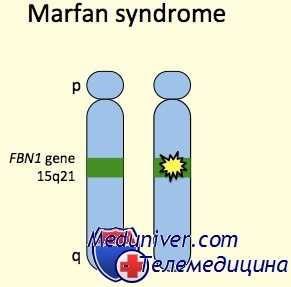
Этиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797).
Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье.
Патогенез синдрома Марфана
Ген FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже.
Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл.
Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность.
Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи).
В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы.
Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb.
Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома Марфана
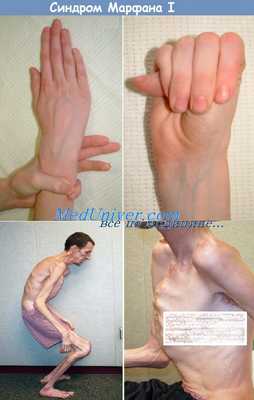
Синдром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов
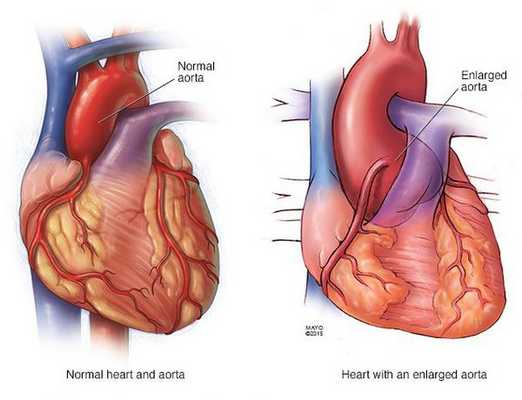
Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе.
Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности.
С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни.
Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин.
Особенности фенотипических проявлений синдрома Марфана:
• Возраст начала: раннее детство
• Несоразмерно высокий рост
• Скелетные аномалии
• Эктопия (подвывих) хрусталика
• Пролапс митрального клапана
• Аневризма и разрыв аорты
• Спонтанный пневмоторакс
• Грыжа оболочки мозга в пояснично-крестцовом отделе
Лечение синдрома Марфана
Синдром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу.
Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая.
Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях.
Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов.
Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью.
Риски наследования синдрома Марфана
Пациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1.
Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было.
При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Наследственные аритмии. Лечение сердечных нарушений синдрома Марфана
У некоторых больных развиваются выраженные желудочковые и наджелудочковые аритмии. Причем если наджелудочковые аритмии часто сопутствуют хронической митральной регургитации, то желудочковые аритмии обычно встречаются при пролапсе митрального клапана и могут достигать такой тяжелой степени, что их с трудом удается подавить.
У некоторых пациентов с пролапсом митрального клапана наблюдают синдром автономной дисфункции, атипичную боль в груди и сердцебиение, не связанные с выраженными аномалиями соединительной ткани.
Иногда у больных с синдромом Marfan без клинически значимых аномалий клапанного аппарата развивается умеренная или выраженная дисфункция ЛЖ. Хотя такую ситуацию можно оценивать как маловероятное совпадение синдрома Marfan и идиопатической дилатационной кардиомиопатии (КМП), было высказано предположение, что снижение функции миокарда может быть связано с некоторыми мутациями гена фибриллина — прямо или опосредованно через TGF-бета. Были получены данные как подтверждающие, так и опровергающие эту гипотезу. Дальнейшие исследования должны прояснить этот вопрос.
Стандартные ведение больных и лечение кардиологических нарушений при синдроме Marfan многогранны: это регулярное клиническое и ЭхоКГ обследование, профилактика эндокардита при проведении стоматологических и других оперативных вмешательств; ограничение подъема тяжестей, контактных видов спорта и любых усилий с максимальным напряжением (при которых возможны телесные повреждения). В качестве базисной терапии применяют длительное лечение Р-АБ, при необходимости — с индивидуальными вариантами.
Подтверждение роли антагонистов бета-адренергических рецепторов было получено в ряде проспективных исследований, показавших снижение скорости дилатапии аорты и риска ее расслоения у больных, принимавших пропранолол или атенолол в дозах, вызывающих отрицательный ипотропный эффект. Кратковременное применение пропранолола у больных с аневризмой с увеличенным синусом Valsalva снижало ЧСС и пиковое САД, однако не улучшало характеристик импеданса, полученных на восходящей аорте.
В то же время, если учесть результаты исследований, показавших значимость центрального пульсового давления при дилатации аорты, то использование бета-АБ представляется оправданным. На мышиной модели синдрома Marfan было показано, что блокатор рецептора апгиотензина 1-го типа лозартан предотвращал появление аномалии стенки аорты, если лечение начинали с рождения, и вызывал регрессию патологических проявлений, включая дилатацию, если лечение начинали в возрасте 2 месяцев. В настоящее время продолжается клиническое исследование этого препарата с участием детей и подростков с выраженной дилатацией корня аорты.
Женщины с синдромом Marfan должны учитывать два момента в связи с возможной беременностью. Первый — риск наследования ребенком заболевания равен 50% (в ряде случаев проводят пренатальную диагностику). Второй — риск расслоения аорты в период беременности, обусловленный гемодинамическими стрессами.
В нескольких десятках историй болезни подтверждается повышенная частота расслоения аорты в III триместре беременности, при родах и в первые месяцы после родов. В то же время в большинстве случаев имеет место выраженная дилатация аорты. Проспективное наблюдение за 21 женщиной в ходе 45 беременностей подтвердило сделанное ранее заключение об относительно низком сердечпо-сосудистом риске, если диаметр аорты не превышает 40 мм и функция сердца не нарушена; эту точку зрения разделяет большинство исследователей. При синдроме Marfan повышается вероятность осложнений во время родов у матери и плода.
- Вернуться в оглавление раздела "Кардиология"
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Аномалии сосудов. Наследственные заболевания соединительной ткани - синдром MARFAN
Одни генетически детерминированные нарушения поражают преимущественно артерии (некоторые касаются только аорты), другие поражают вены и лимфатические сосуды. При некоторых аномалиях нарушается развитие нескольких типов сосудов.
Заболевания соединительной ткани (дисплазии) подразделяют на две большие группы: (1) болезни, вызываемые мутациями в одном гене, которые детерминируют повреждение или каким-то образом повреждают компоненты внеклеточного матрикса; (2) болезни, вызываемые внешними факторами, поражающими внеклеточный матрикс, например ревматоидный артрит и системная красная волчанка. К первой группе относят и многие заболевания с поражением ССС. Подверженность к так называемым приобретенным заболеваниям соединительной ткани отчасти определяется генетически.
Некоторые из них служат причиной развития различных артериопатий. Синдром Williams — наследственное заболевание, вызываемое делецией генов в хромосоме 7q 11.23, включая и тот, который кодирует тропоэластии (ELN), описан далее. Наследуемый по аутосомно-доминантному типу надклаианный стеноз аорты, который проявляется такой же артериопатией, как и синдром Williams, ассоциирован с мутациями, затрагивающими только ELN.
При некоторых заболеваниях соединительной ткани кажется, что патология сосудов связана с ослаблением их стенки, например ломкость артерий, наблюдаемая при сосудистой форме синдрома Ehlers-Danlos, вызываемого мутациями гена, кодирующего проколлаген типа III, COI.3A1. Похожие патогенетические механизмы вызывают, вероятно, развитие атипичных случаев аневризмы аорты или пролапса клапана, наблюдаемых у больных с классической формой синдрома Ehlers-Danlos и несовершенным остеогенезом.
В то же время нарушения ССС, наблюдаемые при синдроме Marfan, вызванном мутациями гена, кодирующего фибриллин-1 (гликопротеин внеклеточного матрикса), не связаны со «слабостью» соединительной ткани. На оборот, большинство фенотииических особенностей обусловлено нарушениями регуляции активности трансформирующего фактора роста бета (ТСЕ-бета), которая обычно контролируется при связывании с фибриллином. Другие наследуемые по законам Менделя артериопатий, как было недавно показано, вызваны дефектами рецепторов TGF-P.
Синдром MARFAN
Это аутосомно-доминантное заболевание встречается довольно часто (2-3 случая на 10 тыс. чел.) среди представителей всех рас и этнических групп. Несмотря на установление генетических и биохимических основ заболевания, диагностика синдрома Marfan вне семей с классическими фенотипическими проявлениями базируется в основном на анализе клинических симптомов. Современные критерии разработаны с учетом пораженных органов и систем: органов зрения, скелета, сердца и аорты, других систем организма, а также семейного анамнеза.
Наличие более специфичных для синдрома Marfan проявлений, таких как дилатация аорты, расслоение аорты у молодых людей без АГ, эктопия хрусталика и эктазия твердой мозговой оболочки, очевидно, более важно с точки зрения диагностики, чем нарушения, характерные для других заболеваний соединительной ткани и встречающиеся в популяции в целом, например сколиоз, гипермобильность суставов, миопия и пролапс митрального клапана (ПМК).
К наиболее характерным нарушениям со стороны ССЗ относятся ПМК и дилатация синусов Valsalva. Клинические проявления этих нарушений в виде митральной peгургитации, аортальной регургитации и расслоения аорты и, при отсутствии лечения, становятся причиной большинства случаев ранней смерти, в результате средний возраст таких пациентов < 40-50 лет. У детей наблюдается тенденция к более тяжелым поражениям митрального клапана, тогда как поражения аорты прогрессируют и наиболее вероятны в подростковом и более старшем возрастах.
Синдром Марфана - симптомы и лечение
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 7 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
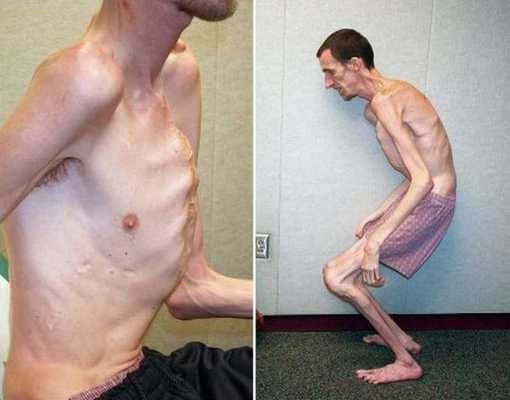
Причины синдрома Марфана
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Тип наследования синдрома — аутосомно-доминантный. Для болезни характерна высокая пенетрантность (частота появления гена) и различная экспрессивность. [5]
Соотношение представителей мужского пола и женского одинаковое.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Марфана
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
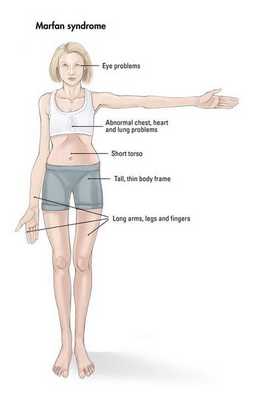
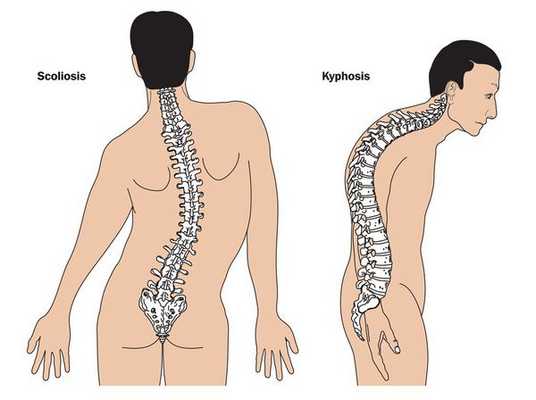
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
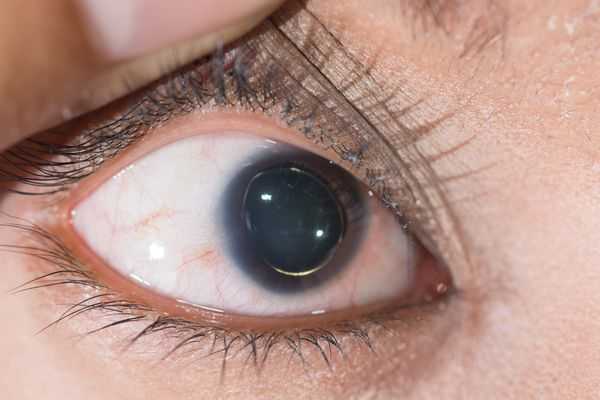
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
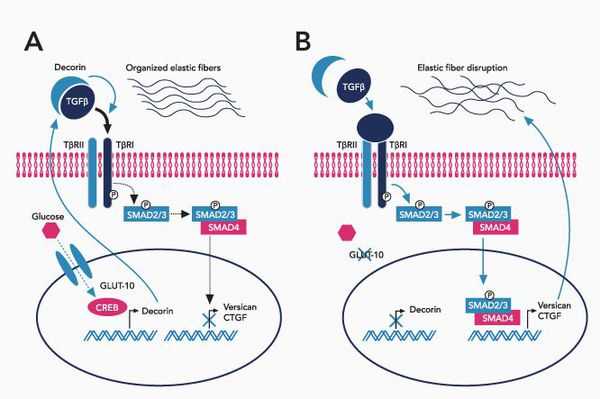
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. [7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. [4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
Классификация и стадии развития синдрома Марфана
Код синдрома Марфана по Международной классификации болезней (МКБ-10): Q87.4.
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
Осложнения синдрома Марфана
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
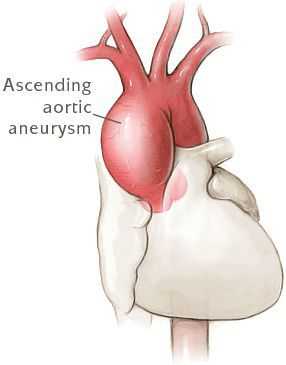
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
Диагностика синдрома Марфана
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
Синдром Марфана
Синдром Марфана - дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
МКБ-10
Общие сведения
Синдром Марфана - системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана - одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста - TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных - первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую - со слабо выраженными изменениями в 1-2-х системах
- выраженную - со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек - соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана - миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца - коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ - обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов - выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Читайте также:
- Лучевая диагностика врожденного отсутствия сегментов печени
- Оригинальный сумамед и его аналоги. - В чем различие?
- Лекарства и электрокардиостимулятор. Взаимодействие антиаритмических препаратов и ЭКС
- Ложная кишечная непроходимость на УЗИ. УЗИ диагностика лихорадки неясного генеза.
- Прогрессия доброкачественных опухолей. Промежуточные стадии развития опухоли.