Поздняя кожная порфирия
Добавил пользователь Alex Обновлено: 29.01.2026
Поздняя кожная порфирия – заболевание, при котором на коже под воздействием солнечного света образуются пузырьки; наиболее распространенная форма порфирии.
Поздняя кожная порфирия встречается во всем мире и является единственной ненаследственной формой порфирии.
На подвергающихся воздействию солнца участках кожи, например внутренних поверхностях рук, кистях и лице, возникает сыпь. Кожа, особенно на руках, легко травмируется. На месте элементов сыпи образуются струпы, затем рубцевание; струпы заживают долго. Повреждение кожи возникает, поскольку порфирины, образованные в печени, переносятся кровью к коже. Может усилиться рост волос на лице.
Часто из-за инфекции вирусом гепатита C или в результате чрезмерного употребления алкоголя повреждается печень. Через некоторое время могут развиваться цирроз и даже злокачественные опухоли печени.
- При возникновении пигментации или пузырей на открытых участках тела.
- При повышенной травматизации кожи на руках.
- При усилении роста волос на лице.
- При увеличении и болезненности печени.
- При покраснении ладоней, появлении «печеночных звездочек» на коже.
Поздняя кожная порфирия (печеночная порфирия) возникает, если уропорфириногендекарбоксилаза, один из ферментов печени, необходимый для синтеза гема, становится неактивным. К провоцирующим факторам относятся железо, алкоголь, эстрогены и инфекция вирусом гепатита C. Реже поздняя кожная порфирия возникает у людей, инфицированных вирусом иммунодефицита человека (ВИЧ). Хотя заболевание ненаследственное, иногда наследуется частичный (от одного родителя) дефицит уропорфириногендекарбоксилазы, что делает человека склонным к развитию заболевания. В таких случаях болезнь называется семейной кожной порфирией.
Чтобы диагностировать позднюю кожную порфирию, плазму крови, мочу и кал проверяют на наличие порфиринов. Любая порфирия, которая вызывает повреждение кожи, сопровождается высоким содержанием порфиринов в плазме крови. При поздней кожной порфирии, кроме того, увеличено содержание порфиринов в моче и кале.
Поздняя кожная порфирия хорошо поддается лечению. Чаще всего прибегают к так называемой венотомии, при которой каждые 1-2 недели удаляют 1 л крови. Это вызывает у пациента небольшой недостаток железа. Содержание порфиринов в печени и плазме крови постепенно снижается, состояние кожи улучшается и в конечном счете полностью нормализуется. Обычно требуется всего 5-6 венотомий; если проводится слишком много процедур, развивается анемия. Дополнительные венотомии необходимы, только если заболевание рецидивирует.
Эффективен прием очень низких доз хлорохина (делагила). Это лекарство выводит избыточные порфирины из печени. Однако высокие дозы хлорохина (даже традиционно используемые при лечении других болезней) приводят к слишком быстрому выведению порфиринов, что вызывает временное ухудшение течения заболевания и повреждение печени. Рекомендуют прекратить употребление алкоголя.
Часто из-за инфекции вирусом гепатита C или в результате чрезмерного употребления алкоголя повреждается печень. Через некоторое время могут развиваться цирроз и даже злокачественные опухоли печени.
Поздняя кожная порфирия
Поздняя кожная порфирия является наиболее распространенной формой порфирии. Это наиболее частая форма болезни с наследственной предрасположенностью, для реализации которой требуются дополнительные токсические факторы внешней среды. В статье освещены вопросы этиологии и патогенеза поздней кожной порфирии, представлены клинические проявления данного заболевания. По современным воззрениям, эта разновидность порфирии является многофакторным заболеванием, обусловленным латентным или манифестным повреждением печени с последующим нарушением порфиринового обмена. В патогенезе имеют значение разнообразные экзогенные и эндогенные факторы. В статье приведена классификация клинических проявлений поздней кожной порфирии. В зависимости от преобладания поражения кожи, внутренних органов или нервной системы, выделяют 4 клинические формы поздней кожной порфирии: кожную, кожно-висцеральную, кожно-нервную и смешанную. У 80% больных наблюдаются атипичные формы: склеродермоподобная, язвенно-некротическая и меланодермическая. Диагностика различных форм наследственных эритропоэтических и печеночных порфирий основывается на характерных лабораторных данных и изменениях кожи. В статье освещены основные диагностические критерии поздней кожной порфирии, приведены патогномоничные симптомы. Представленный клинический случай демонстрирует диагностику поздней кожной порфирии у пациентки с ВИЧ-инфекцией.
Ключевые слова: поздняя кожная порфирия, многофакторное заболевание, ВИЧ-инфекция, порфирины.
Для цитирования: Уфимцева М.А., Канева Е.В., Худорожкова Н.П. Поздняя кожная порфирия // РМЖ. Дерматология. 2016. № 10. С. 651–654.
Для цитирования: Уфимцева М.А., Канева Е.В., Худорожкова Н.П. Поздняя кожная порфирия. РМЖ. 2016;10:651-654.
Porphyria cutanea tarda
+Ufimtseva M.A., Kaneva E.V., Khudorozhkova N.P.
Ural State Medical University, Ekaterinburg, Russia
Porphyria cutanea tarda is the most prevalent subtype of porphyria and the most common inherited condition produced by additional exogenous factors. The paper reviews etiology and pathogenesis as well as clinical signs of porphyria cutanea tarda. Currently, this subtype of porphyria is considered as a multifactorial disorder which is resulted from the latent or manifest liver damage followed by porphyrin metabolism abnormalities. A variety of exogenous and endogenous factors contribute to the pathogenesis of this disease. The paper addresses the classification of clinical manifestations of porphyria cutanea tarda. It is classified by the prevalent site of lesion into cutaneous, cutaneous visceral, cutaneous nervous, and mixed subsets. 80% of these patients present with atypical forms, i.e., scleroderma-like, ulcero-necrotic, or melanoderma-like. Inherited erythropoietic and hepatic porphyrias are diagnosed by typical abnormalities in laboratory tests and skin manifestations. Major diagnostic criteria of porphyria cutanea tarda and pathognomonic symptoms are discussed. The clinical case illustrates the diagnosis of porphyria cutanea tarda in an HIV-positive woman.
Key words: HIV-positive patient, multifactorial disorder, HIV infection, porphyrins.
For citation: Ufimtseva M.A., Kaneva E.V., Khudorozhkova N.P. Porphyria cutanea tarda // RMJ. Dermatology. 2016. № 10. P. 651–654.
Статья посвящена проблеме поздней кожной порфирии
Определение, эпидемиология
Поздняя кожная порфирия (ПКП) – наиболее распространенная форма порфирии, обусловленная нарушением синтеза гемов печени, сопровождающаяся повышенным образованием и выделением уропорфирина и копропорфирина мочой и задержкой их в коже [1].
Выявляется с частотой 0,5 случая на 100 тыс. населения [1, 2]. Как правило, болеют мужчины в возрасте 40–50 лет [2–4]. В то же время за минувшие 10 лет в 3 раза в сравнении с 1970-ми годами увеличилось число женщин с ПКП [2].
ПКП – заболевание с хроническим рецидивирующим течением, четко связанным с сезонами года. Рецидивы болезни у 90–95% больных наступают в весенне-летний период [5].
Этиология
Многочисленные исследования подтверждают, что порфирия редко зависит от одной определенной причины, но чаще является многофакторным заболеванием и набор факторов риска индивидуален для каждого пациента [6].
Выделяют 2 основных типа заболевания:
1) наследственный (аутосомно-доминантный), выявляется в детском возрасте во всех поколениях;
2) приобретенный (спорадический), при котором у всех без исключения больных имеет место сочетанная патология внутренних органов:
а) стеатогепатит (алкогольной и/или вирусной этиологии);
б) патология сердечно-сосудистой системы (артериальная гипертензия, стенокардия, инфаркт миокарда в анамнезе);
в) патология ЖКТ (язвенная болезнь желудка и двенадцатиперстной кишки, хронический панкреатит алкогольной этиологии);
г) патология дыхательной системы (хронический обструктивный бронхит), курение;
д) сахарный диабет;
е) хронические дерматозы (диссеменированная красная волчанка, хроническая пиодермия, экзема, псориаз).
Ведущим фактором в развитии ПКП является употребление алкоголя. При наличии наследственной предрасположенности даже кратковременная интоксикация способна привести к быстрому прогрессированию специфических нарушений порфиринового обмена. В то же время при отсутствии генетически детерминированных дефектов в системе ферментов поломка в метаболизме порфиринов возникает после продолжительной интоксикации и обусловлена прямым токсическим действием [1, 4, 7]. Еще одним немаловажным триггерным фактором является наличие хронической НСV-инфекции. Активно протекающий хронический гепатит С диагностируют у 25–90% пациентов с ПКП в разных регионах планеты, в том числе в России [8].
Среди страдающих ПКП 66% женщин принимали эстрогены (с этим, возможно, связано увеличение числа больных порфирией женщин за последние 10 лет), 13% исследуемых были ВИЧ-инфицированы [7]. Некоторые исследования предполагают участие ВИЧ-инфекции в повреждении окислительной системы печени, что приводит к нарушению обмена порфирина и таким образом провоцирует развитие порфирии [6, 9–12]. Диагноз тяжелой ПКП, особенно у молодых людей, должен служить причиной немедленного обследования на ВИЧ [11].
Перегрузка железом также зачастую связана с этим заболеванием и, как полагают, играет определенную роль в его патогенезе [3, 13]. Еще одним предрасполагающим фактором развития порфирии является длительный контакт с нефтепродуктами [8, 14, 15].
Патогенез
Блокирование или частичное снижение активности уропорфириноген-III-декарбоксилазы, участвующей в биосинтезе порфиринов, с увеличением активности печеночной синтетазы сигма-аминолевулиновой кислоты приводит к увеличению уровня порфиринов (плазмы, мочи, печени, кала) с последующей фотосенсибилизацией тканей [16, 17].
Классификация
В зависимости от преобладания поражения кожи, внутренних органов или нервной системы выделяют 4 клинические формы поздней кожной порфирии: кожную, кожно-висцеральную, кожно-нервную и смешанную.
Кожная форма характеризуется изолированным поражением кожи при нормальных показателях печеночных проб (трансаминаза, билирубин, щелочная фосфатаза).
Для кожно-висцеральной формы характерно полиорганное проявление заболевания. В развитии патологии провоцирующим фактором служат перенесенные заболевания печени, в том числе вирусный гепатит. Поражения печени развиваются через 1–2 года на фоне кожных проявлений, что выявляется на УЗИ (гепатомегалия).
Кожно-нервная форма поздней кожной порфирии развивается у подавляющего большинства пациентов на фоне черепно-мозговой травмы (ушиб, сотрясение). Сопровождается развитием полиневритического синдрома, который характеризуется гипестезией в области рук и лица, похуданием лица, кистей и плечевого пояса со снижением мышечной силы. Одновременно развиваются вегетативно-трофические нарушения (выраженный гипертрихоз в височно-скуловой области, дистрофические изменения ногтевых пластин, преждевременное старение кожи лица). Реже встречается астено-вегетативный синдром с трофическими нарушениями.
Смешанная форма поздней кожной порфирии развивается у лиц с отягощенным анамнезом (употребление наркотиков, алкоголизм, туберкулез легких, сифилис, малярия, бруцеллез). Характеризуется быстрым развитием поражения нервной системы (полиневритический, астено-вегетативный синдром с выраженными трофическими нарушениями) и патологическими изменениями печени, которые прогрессируют и завершаются формированием цирроза печени [2].
Клиника
Клинически ПКП характеризуется появлением на открытых участках кожи (тыл кистей, лицо, шея, ушные раковины) зудящих пузырей величиной до 10 мм в диаметре, наполненных прозрачным содержимым [2]. Пузыри быстро вскрываются с образованием эрозий, которые покрываются корками, иногда серозно-гнойного характера. Отмечается легкая ранимость кожи при малейшей травме [1].
Высыпания обычно возникают после солнечного облучения, вследствие чего обострения заболевания наблюдаются летом и осенью при солнечной погоде. Постепенно нарастает меланоз открытых участков кожи, усиливаются естественные складки лица, появляются морщины, признаки преждевременного старения кожи. Больные выглядят значительно старше своих лет. Наблюдается фиолетово-коричневое окрашивание век и конъюнктивы. На месте бывших пузырей остаются атрофические поверхностные рубцы.
ПКП свойственны поражения печени, нарушение функций ЖКТ, ожирение. Встречаются различные поражения органа зрения: конъюнктивит, расширение сосудов глазного дна, помутнение роговицы, нарушение цветовосприятия [16, 18–20].
При лабораторных исследованиях выявляется повышение активности аминотрансфераз, гамма-глутамилтрансферазы, гипергаммаглобулинемия, гиперпорфиринемия, гиперферремия. Патогномонично увеличение концентрации уропорфирина и копропорфирина I и III фракций [16, 20, 21].
Клинический случай
Пациентка Д., 36 лет, считает себя больной с апреля 2012 г., когда, проснувшись утром, заметила два пузырька на носу размером 2–3 мм, с прозрачным содержимым. Самостоятельно вскрыла, после чего образовались корки желтоватого цвета, наступил полный регресс кожных высыпаний.
Второй эпизод заболевания наблюдался в июле 2012 г.: пузырьки 3–4 мм на тыльной поверхности обеих кистей, с прозрачным содержимым. Обратилась за помощью к дерматологу по месту жительства, где был поставлен диагноз: фотодерматоз. Получала лечение: Sol. Chloropyramini 2% – 1 ml № 5 in amp., внутримышечно; Sol. Calcii chloridi 10% – 10 ml № 10 in amp., внутривенно; Sol. Betamethasoni 1,0 ml № 1 in amp., внутримышечно. На фоне проводимого лечения стали появляться новые пузыри размером от 3 до 10 мм, с тенденцией к периферическому росту. На месте пузырей образовались медленно заживающие, резко болезненные эрозии.
После проведения дополнительного обследования, на основании оранжево-красного свечения мочи под лампой Вуда поставлен диагноз: поздняя кожная порфирия. Далее произведена коррекция лечения: Caps. Tocopheroli acetatis 0,2 № 20, по 1 капсуле 1–2 раза в сутки в течение 1 нед.; Ung. Dioxomethyltetrahydropyrimidini+Chloramphenicoli 100,0, наружно на высыпания утром; Linimenti Zinci oxydi 10% – 30 ml, тонким слоем на места поражения на ночь, для лица – фотозащитный крем с SPF 50. Положительного эффекта от терапии не наблюдалось. Пузыри продолжали появляться на лице, шее, верхних конечностях и пальцах стоп.
В анамнезе: BИЧ IVБ стадии с 2000 г., фаза мнимой ремиссии на фоне ретровирусной терапии; в 2002 г. – аппендэктомия. Употребление героина в 1993–1995 гг. Ранее – алкоголизм, на данный момент употребление алкоголя 1 раз в месяц 0,5 л водки. Курение с 1998 г., 1 пачка сигарет в день.
Локальный статус: кожа вне очагов высыпаний физиологической окраски, сухая, тургор снижен. Кожный процесс носит распространенный характер, симметричный, локализован на лице, шее, верхних конечностях, преимущественно на тыльной поверхности кистей, кожи стоп. Процесс представлен везикулами, пузырями с серозным, серозно-геморрагическим содержимым, покрышка напряжена, легко травмируется. На месте вскрывающихся везикул – эрозии ярко-красного цвета, серозно-геморрагические и серозно-гнойные корки. Кожа в местах поражения гиперемирована, утолщена. В местах заживших дефектов отмечаются очаги дисхромии с нечеткими границами и атрофические рубцы неправильных очертаний (рис. 1). Субъективно: чувство жжения, умеренная болезненность эрозий. Общее состояние удовлетворительное: предъявляет жалобы на слабость, общее недомогание.
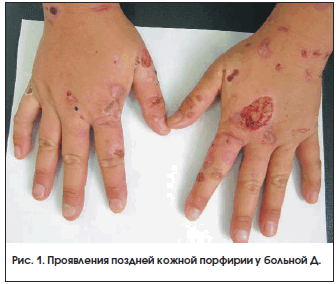
Моча в свете ультрафиолетовых лучей лампы Вуда имеет оранжево-красное свечение. В биохимических анализах крови: повышение АСТ до 51,22 (референсный интервал 5–34 МЕ/л), глобулинов – до 34,07 (19–30 г/л), снижение альбуминов до 34,08 (35–54 г/л). Общий анализ крови: СОЭ – 30 мм/ч, остальные показатели в пределах нормы. Анализ крови на маркеры вирусных гепатитов – отрицательный результат. Микроскопическое исследование на клетки Тцанка и эозинофилию пузыря – отрицательный результат. Исследование кала на гельминты и простейшие – не обнаружены. Содержание уропорфиринов в моче – в норме, копропорфирина – повышено. Порфирины в кале не определялись. Антитела к Treponema pallidum не обнаружены. Другие лабораторные показатели в пределах нормы. Патологии со стороны других органов и систем не выявлено.
Лечение: Disol 400 ml, внутривенно капельно (Натрия ацетат + Натрия хлорид); Tab. Calcii gluconatis 0,5 № 40, по одной таблетке внутрь 3 раза в день; Tab. Cetirizini 0,05 № 10, по одной таблетке внутрь 1 раз в день; Sol. Fucorcinum 10 ml, наружно на очаги поражения 2–4 раза в сутки; Ung. Betamethasoni + Gentamycini 30,0, наружно на очаги поражения 2 раза в сутки.
Проводимую терапию перенесла удовлетворительно, без побочных эффектов. Состояние улучшилось. Высыпания на всех участках пораженной кожи перешли в стадию регресса, на местах инволюции элементов – очаги дисмеланоза. Прогноз благоприятный.
1. Оркин В.Ф., Шабогина А.А., Олехнович Н.М., Платонова А.Н., Надежкина Е.В., Кочнева Е.В. Поздняя кожная порфирия // KlinDermatolVenerol. 2008. № 3. С. 20–23 [Orkin V.F., Shabogina A.A., Olehnovich N.M., Platonova A.N., Nadezhkina E.V., Kochneva E.V. Pozdnjaja kozhnaja porfirija // KlinDermatolVenerol. 2008. № 3. S. 20–23 (in Russian)].
2. Кузнецова Н.П., Чащин А.Ю., Афанасьева И.Г. Отдаленные результаты лечения и реабилитации больных поздней кожной порфирией // Рос. журн. кож. и вен. бол. 2007. № 5. С. 8–11 [Kuznecova N.P., Chashhin A.Ju., Afanas'eva I.G. Otdalennye rezul'taty lechenija i reabilitacii bol'nyh pozdnej kozhnoj porfiriej // Ros. zhurn. kozh. i ven. bol. 2007. № 5. S. 8–11 (in Russian)].
3. Кривошеев А.Б., Кривошеев Б.Н., Кондратова М.А. Метаболический синдром и поздняя кожная порфирия // Рос. журн. кож. и вен. бол. 2011. № 5. С. 20–27 [Krivosheev A.B., Krivosheev B.N., Kondratova M.A. Metabolicheskij sindrom i pozdnjaja kozhnaja porfirija // Ros. zhurn. kozh. i ven. bol. 2011. № 5. S. 20–27 (in Russian)].
4. Кривошеев А.Б., Кривошеев Б.Н., Морозов Д.В. Латентная поздняя кожная порфирия: клинические, биохимические и прогностические аспекты // Рос. журн. кож. и вен. бол. 2008. № 3. С. 8–15 [Krivosheev A.B., Krivosheev B.N., Morozov D.V. Latentnaja pozdnjaja kozhnaja porfirija: klinicheskie, biohimicheskie i prognosticheskie aspekty // Ros. zhurn. kozh. i ven. bol. 2008. № 3. S. 8–15 (in Russian)].
5. Кривошеев Б.Н., Кривошеев А.Б. Биоритмы метаболизма порфиринов при поздней кожной порфирии // Рос. журн. кож. и вен. бол. 2007. № 2. С. 37–42 [Krivosheev B.N., Krivosheev A.B. Bioritmy metabolizma porfirinov pri pozdnej kozhnoj porfirii // Ros. zhurn. kozh. i ven. bol. 2007. № 2. S. 37–42 (in Russian)].
6. Egger N.G., Goeger D.E., Payne D.A. et al. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency // Dig. Dis. Sci. 2002. Vol. 47. P. 419–426.
7. Jalil S., Grady J.J., Lee C., Anderson K.E. Associations among behavior-related susceptibility factors in porphyria cutanea tarda // Clin. Gastroenterol. Hepatol. 2010. Vol. 8. P. 297–302.
8. Кривошеев А.Б., Кривошеев Б.Н. Противовирусная терапия поздней кожной порфирии, ассоциированной с хроническим вирусным гепатитом С // Рос. журн. кож. и вен. бол. 2009. № 2. С. 39–45 [Krivosheev A.B., Krivosheev B.N. Protivovirusnaja terapija pozdnej kozhnoj porfirii, associirovannoj s hronicheskim virusnym gepatitom S // Ros. zhurn. kozh. i ven. bol. 2009. № 2. S. 39–45 (in Russian)].
9. Almehni A., Deliri H., Szego G.G. et al. Porphyria cutanea tarda in a patient with HIVinfection // W. V. Med. J. 2005. Vol. 101. P. 19–21.
10. Aziz Ibrahim A., Esen U.I. Porphyria cutanea tarda in pregnancy: a case report // J. Obstet. Gynaecol. 2004. Vol. 24. P. 574–575.
11. Hift R.J., Corrigall A.V., Hancock V. et al. Porphyria cutanea tarda: the etiological importance of mutations in the HFE gene and viral infection is population-dependent // Cell. Mol. Biol. (Noisy-le-grand). 2002. Vol. 48. P. 853–859.
12. Wickliffe J.K., Abdel-Rahman S.Z., Lee C. et al. CYP1A2*1F and GSTM1 alleles are associated with susceptibility to porphyria cutanea tarda // Mol. Med. 2011. Vol. 17. P. 241–247.
13. Labidi J. Porphyria cutanea tarda in a chronic hemodialysis patient // Saudi J. Kidney Dis. Transpl. 2010. Vol. 21. P. 919–922.
14. Васильев О.Н. Применение делагила в комплексе с преднизолоном при урокопропорфирии (поздней кожной порфирии) // Рос. журн. кож. и вен. бол. 2005. № 5. С. 38–40 [Vasil'ev O.N. Primenenie delagila v komplekse s prednizolonom pri urokoproporfirii (pozdnej kozhnoj porfirii) // Ros. zhurn. kozh. i ven. bol. 2005. № 5. S. 38–40 (in Russian)].
15. Кривошеев Б.Н., Кривошеев А.Б. Поздняя кожная порфирия как внепеченочное проявление хронической HCV-инфекции // Рос. журн. кож. и вен. бол. 2008. № 2. С. 13–18 [Krivosheev B.N., Krivosheev A.B. Pozdnjaja kozhnaja porfirija kak vnepechenochnoe projavlenie hronicheskoj HCV-infekcii // Ros. zhurn. kozh. i ven. bol. 2008. № 2. S. 13–18 (in Russian)].
16. Кожные и венерические болезни. Справочник / под ред. О.Л. Иванова. М.: Медицина, 1997. С. 213–214 [Kozhnye i venericheskie bolezni. Spravochnik / pod red. O.L. Ivanova. M.: Medicina, 1997. S. 213–214 (in Russian)].
17. Хэбиф Т.П. Кожные болезни. Диагностика и лечение: пер. с англ. Т.П. Хэбиф / под ред. А.А. Кубановой. М.: Медпресс-информ, 2006. С. 376–380 [Hjebif T.P. Kozhnye bolezni. Diagnostika i lechenie: per. s angl. T.P. Hjebif / pod red. A.A. Kubanovoj. M.: Medpress-inform, 2006. S. 376–380 (in Russian)].
18. Кузнецова Н.П., Панков Б.С., Чубарова А.С. и др. Порфирии. М., 1981. С. 66–146 [Kuznecova N.P., Pankov B.S., Chubarova A.S. i dr. Porfirii. M., 1981. S. 66–146 (in Russian)].
19. Монахов С.А. Поздняя кожная порфирия // Рос. журн. кож. и вен. бол. 2002. № 5. С. 83–85 [Monahov S.A. Pozdnjaja kozhnaja porfirija // Ros. zhurn. kozh.-ven. bol. 2002. № 5. S. 83–85 (in Russian)].
20. Пономарев А.А., Куликов Е.П., Караваев Н.С., Федосеев А.В. Редкие кожно-висцеральные синдромы. Рязань, 1998. С. 422–442 [Ponomarev A.A., Kulikov E.P., Karavaev N.S., Fedoseev A.V. Redkie kozhno-visceral'nye sindromy. Rjazan', 1998. S. 422–442 (in Russian)].
21. Фицпатрик Т., Джонсон Р., Вульф К. и др. Дерматология: Атлас-справочник : пер. с англ. М., 1999. С. 262–267 [Ficpatrik T., Dzhonson R., Vul'f K. i dr. Dermatologija: Atlas-spravochnik : per. s angl. M., 1999. S. 262–267 (in Russian)].
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Порфирия
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Порфирия: причины появления, симптомы, диагностика и способы лечения.
Определение
Порфирии (от греч. porphyreis – пурпурный) – это ряд заболеваний обмена веществ, при которых нарушается образование гема, представляющего собой комплексное соединение порфиринов с двухвалентным железом. В результате в организме происходит накопление порфиринов (пигментов, представляющих собой производные порфина) или их предшественников. К счастью, эти патологии встречаются относительно редко ‒ не более 7-12 случаев на 100 000 человек.
Причины появления порфирии
В подавляющем большинстве случаев причиной порфирий становятся генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в синтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается на фоне заболеваний печени (алкогольного гепатита, вирусного гепатита С) или длительной интоксикации тяжелыми металлами.
Наследование порфирий происходит по аутосомно-доминантному (наследуется одна нормальная и одна измененная копия гена, причем измененная копия доминирует и «подавляет» нормальную, в результате чего развивается генетическое заболевание) или аутосомно-рецессивному типу (болезнь Гюнтера). Синтез гема проходит 8 последовательных этапов, за каждый из которых отвечает конкретный фермент, кодируемый определенным геном. Соответственно, для каждой формы порфирии существует специфичный ферментативный дефект.
Наибольшее количество гемов образуется в печени и костном мозге. В печени гемы входят в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и «обезвреживании» различных ксенобиотиков (чужеродных химических веществ, попадающих в организм извне). В костном мозге гемы необходимы для синтеза гемоглобина.
Результатом снижения активности ферментов становится торможение синтеза гемов, что ведет к накоплению его токсичных промежуточных метаболитов.
Провоцирующими факторами развития порфириновой болезни становятся:
- избыточная инсоляция;
- недостаточное питание и скудный рацион;
- систематические стрессы;
- чрезмерное употребление алкоголя;
- вирусные и бактериальные инфекции;
- хронические интоксикации солями тяжелых металлов.
- Беременность.
В отдельных случаях манифестация патологического процесса может произойти на фоне приема некоторых лекарственных препаратов: антибиотиков, антиконвульсантов, нестероидных противовоспалительных средств, пероральных контрацептивов.
Классификация заболевания
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность.
Современная классификация порфирий весьма разветвленная:
I. Эритропоэтические порфирии.
- Эритропоэтическая уропорфирия (врожденная эритропоэтическая порфирия, или болезнь Гюнтера).
- Эритропоэтическая протопорфирия:
- манифестная форма;
- латентная форма.
- Эритропоэтическая копропорфирия.
- Пирролопорфирия (острая перемежающая порфирия, доминантная порфирия шведского типа):
- манифестная форма;
- латентная форма.
- Протокопропорфирия (варигатная, или смешанная порфирия, доминантная порфирия Южно-Африканского типа):
- кожная форма;
- острая форма без кожных проявлений;
- комбинированная форма с кожными и острыми проявлениями;
- латентная форма.
- Урокопропорфирия (поздняя кожная порфирия):
- манифестная приобретенная (симптоматическая) форма (развивается при интоксикациях гексахлорбензолом, опухолях печени и других патологических состояниях;
- манифестная наследственная форма (развивается у лиц с наследственной предрасположенностью);
- латентная наследственная форма (выявляется у родственников больных).
- Наследственная копропорфирия.
- Неклассифицированная печеночная порфирия.
- Гепатоэритропоэтическая порфирия.
- Неклассифицированная печеночная порфирия, протекающая с клиническим синдромом световой оспы.
- Патологические состояния, сопровождающиеся геморрагическим синдромом и нарушениями порфиринового обмена печеночного типа.
- избыточная инсоляция;
- недостаточное питание и скудный рацион;
- систематические стрессы;
- чрезмерное употребление алкоголя;
- вирусные и бактериальные инфекции;
- хронические интоксикации солями тяжелых металлов.
- проба Эрлиха;
- исследование концентрации ферментов в крови;
- ПЦР-тесты на гепатит;
- молекулярно-генетические исследования.
- Наследственная. Развивается вследствие генетической мутации. Наследование происходит по аутосомно-доминантному типу. Снижение уровня уропорфириноген-декарбоксилазы наблюдается в печени, эритроцитах, плазме.
- Спорадическая (приобретенная). Наиболее частая разновидность ПКП. Основные причины – алкоголизм, гепатит С, прием эстрогенов. Низкое содержание уропорфириноген-декарбоксилазы отмечается только в печени.
- Паранеопластическая. Возникает в результате развития гепатомы, продуцирующей избыточные количества порфиринов. Отличительной особенностью является поздний дебют (60-70 лет).
- Псевдо-ПКП. Развивается у больных хронической почечной недостаточностью в терминальной стадии, т. е. находящихся на гемодиализе. Клиническая картина возникает не по причине повышенной продукции порфиринов, а их сниженного выведения. Особенности данной формы – нормальная концентрация порфиринов в моче и очень высокая в плазме, выраженные фототоксические реакции.
- Рутинные лабораторные тесты. В биохимическом анализе крови отмечается увеличение печеночных трансаминаз, билирубина, гамма-глутамилтранспептидазы (специфичного показателя алкогольного поражения печени), ферритина, сывороточного железа, креатинина (при почечной недостаточности), снижение уровня альбумина. При циррозе печени в коагулограмме выявляются признаки гипокоагуляции. У многих пациентов обнаруживаются положительные маркеры вируса гепатита С.
- Специфические лабораторные исследования. Наблюдается повышение концентрации уропорфирина в плазме и моче, копропорфирина в кале. Под лампой Вуда моча принимает красно-оранжевое или ярко-розовое свечение. При люминесцентной микроскопии плазма дает красную флуоресценцию. Снижение активности фермента уропорфириноген-декарбоксилазы в эритроцитах и наличие генетической мутации характерно только для наследственной формы заболевания.
- Инструментальные исследования. Визуализирующие методы (ультразвуковое исследование и КТ брюшной полости) необходимо проводить для исключения злокачественного образования печени, продуцирующего порфирины. Это в первую очередь касается пациентов, у которых симптомы заболевания развились после 60 лет.
Симптомы порфирии
Сроки дебюта рассмотренных выше форм заболевания различны: эритропический тип проявляется в возрасте 3–5 лет, острый печеночный — в 14–16 лет, хронический печеночный — после 40 лет. После манифестации патологии пациенты сталкиваются со специфической симптоматикой.
При острых формах порфирии больные жалуются на сильные боли в животе, задержку стула, учащенное сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого).
Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью и снижением чувствительности по всему телу, прогрессирующей мышечной слабостью, судорожными припадками, различными психическими расстройствами (тревожностью, психомоторным возбуждением, бредом).
При поздней кожной форме на участках кожного покрова, подвергающихся постоянному воздействию солнечного света, формируется гиперпигментация, в результате чего кожа приобретает землистый или бронзовый оттенок.
Выраженными симптомами этой формы порфирии являются везикулезные и буллезные высыпания, которые покрыты корками, шелушение кожи, медленно заживающие эрозии, милиумы.
Кроме того, могут отмечаться явления гипертрихоза (избыточного оволосения) лобно-височной области лица, фотосенсибилизация.
При эритропоэтических порфириях наблюдаются признаки светочувствительности, причем даже более отчетливые, чем при поздней кожной порфирии. Пациент испытывает сильную боль, если на кожный покров попадают прямые солнечные лучи. Обширные эрозии оставляют после себя атрофичексие рубцы, что приводит к обезображиванию внешнего вида больного. В результате множественных атрофических рубцов на коже кистей рук развиваются контрактуры. Избыточное количество порфиринов приводит к тому, что зубная эмаль приобретает красновато-коричневый цвет (эритродонтия), моча становится красной или розовой.
Специфический признак эритропоэтической порфирии – утолщение, огрубение и уплотнение кожи периоральной и периорбитальной зон, крыльев и спинки носа, тыльной поверхности кистей.
Диагностика порфирии
При постановке диагноза учитывается наличие данного заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, беременность).
- Общеклинический № и биохимический анализ крови: АЛТ, АСТ, непрямой билирубин, сывороточное железо, ферритин.
Синонимы: Общий анализ крови, ОАК. Full blood count, FBC, Complete blood count (CBC) with differential white blood cell count (CBC with diff), Hemogram. Краткое описание исследования Клинический анализ крови: общий.
Порфирия

Порфирии — группа генетически обусловленных патологий, проявляющихся на фоне нарушения синтеза гема (промежуточного продукта метаболизма гемоглобина) и накопления его токсичных соединений в организме человека. Симптоматика заболеваний разнообразна: пациенты могут сталкиваться со светочувствительностью, высыпаниями на коже, хроническими болями в животе, частичным или полным параличом, острыми психозами. Диагностика метаболического расстройства осуществляется посредством молекулярно-генетических тестов и лабораторных анализов биоматериалов, полученных врачами от ребенка или взрослого. Медикаментозная терапия направлена на снижение концентрации токсических метаболитов в крови пациента. Хирургические вмешательства выполняются при осложненном течении патологии.
Общая информация
Порфириновая болезнь диагностируется сравнительно редко: российские врачи выявляют не более 12 случаев на 100 тысяч человек. Различные формы патологии получают распространение в отдельных регионах Земли. Так, признаки поздней кожной порфирии часто выявляется у жителей Южной Африки (1 случай на 800 человек). Острый перемежающийся тип заболевания характерен для жителей Северной Европы (1 случай на 1000 человек). Мужчины и женщины одинаково часто страдают от различных форм синтеза гема.
Причины развития
Основным фактором, провоцирующим развитие заболевания, становятся генетические мутации в организме носителя. Единственным исключением остается поздняя кожная порфирия — ее происхождение связано с патологиями печени или длительными интоксикациями человеческого организма солями тяжелых металлов.
Порфириновая болезнь относится к наследуемым метаболическим расстройствам. Отдельные симптомы порфирии или приступы сопровождающих ее психозов могут проявиться в любом возрасте. Специфический набор признаков патологии формируется после преодоления пациентом пубертатного возраста.
Синтез гема состоит из восьми последовательных этапов, протекающих под действием ферментов. Выработка сложных белковых соединений кодируется определенным геном. Порфириновая болезнь приводит к образованию ферментативных дефектов. Разные формы заболевания развиваются на фоне повреждения различных генетических кодов. Гем состоит из порфиринов, объединенных с железом. Продуцирование этих соединений выполняется тканями печени и костного мозга. На основании гема формируются гемоглобин. Нарушение его синтеза приводит к образованию токсичных метаболитов, скапливающихся в тканях человеческого организма.
Провоцирующими факторами манифестации порфириновой болезни становятся:
В отдельных случаях обострение патологии происходит из-за приема пациентами антибиотиков, антиконвульсантов, нестероидных противовоспалительных средств, пероральных контрацептивов.
Этиология
Нарушение химического состава ферментов, участвующих в продуцировании гема, приводит к формированию токсичных для человека метаболитов. Хронические формы порфирии характеризуются накоплением в тканях протопорфирина, копропорфирина и уропорфирина. Острые типы заболевания развиваются на фоне повышенной концентрации в крови порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Накопление порфиринов в кожных покровах детей и взрослых происходит под действием ультрафиолета. Побочным эффектом этого процесса становится окисление липидов. Под действием солнечного света погибают клетки кожи. Копропорфирин провоцирует усиление пигментации дермы и усиленный рост волос. Отложение протопорфирина в печени становится причиной закупорки желчных протоков. Уропорфирины приводят к ускоренной деструкции эритроцитов в селезенке. ДАЛК и порфобилиноген накапливаются в нервных тканях и запускают процесс дегенерации аксонов.
Виды патологии
Врачи используют несколько типологий порфириновой болезни, основанных на клинической симптоматике, локализации нарушений метаболизма порфиринов или вовлеченных тканей. Наиболее часто гематологи выделяют три формы патологии: эритропоэтическую, острую печеночную и хроническую печеночную.
Эритропоэтический тип заболевания характеризуется поражением кожных покровов под действием солнечных лучей. Данная форма патологии рассматривается врачами как наиболее тяжелая.
Острые печеночные виды порфириновой болезни характеризуются приступообразным течением. Основной мишенью токсичных метаболитов гемоглобина становится нервная система. В редких случаях заболевание осложняется светочувствительностью.
Хронические печеночные болезни часто сопровождаются появлением поздней кожной порфирии. ПКП может оказаться наследственной или приобретенной патологией. Врачи рассматривают ее как наиболее благоприятный для лечения вид порфириновой болезни.
Симптомы
Ранние симптомы порфирии неспецифичны. Сроки дебюта рассмотренных выше форм заболевания различны: эритропический тип проявляется в 3–5 лет, острый печеночный — в 14–16 лет, хронический печеночный — после 40 лет. Вслед за манифестацией патологии пациенты сталкиваются со специфической симптоматикой.
Острые симптомы включают боли в животе, задержку стула, резкое повышение частоты сердечных сокращений, рост артериального давления, изменения окраски мочи. Пациент страдает от снижения чувствительности конечностей, ощущения ломоты в костях и суставах. Появляется мышечная слабость, в тяжелых случаях — паралич.
Позднее развиваются судорожные припадки, бред и галлюцинации.
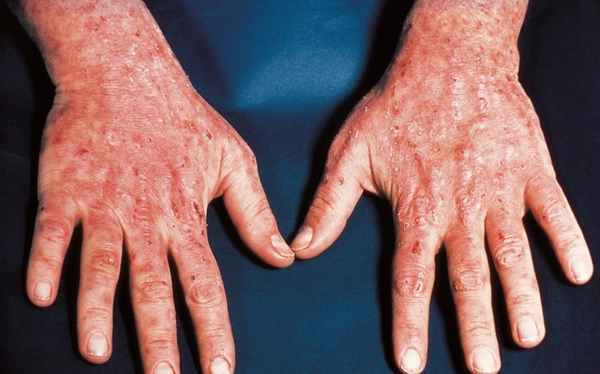
ПКП характеризуется формированием участков гиперпигментации кожных покровов под воздействием солнечного света. Высыпания землистого или бронзового цвета возникают на лице, шее, ушных раковинах и кистях рук. При осложненном течении патологии пациент сталкивается с гипертрихозом: избыточный рост волос локализуется на лобно-височной области лица. На коже появляются пузыри-пустулы с жидким содержимым. Вскрывшийся очаг образует эрозию, после заживления которой образуется атрофический рубец.
Эритропоэтическая форма порфириновой болезни провоцирует развитие у пациента более выраженных признаков светочувствительности в сравнении с ПКП. Ребенок или взрослый испытывает сильную боль при попадании на кожу прямых солнечных лучей. Очаги эрозии занимают значительную площадь и приводят к формированию грубых рубцов, меняющих внешность человека. Рубцовая ткань снижает подвижность локтевых и коленных суставов. Моча пациента обретает красный или розовый оттенок, эмаль зубов становится красно-коричневой. Кожные покровы лица и тыльной поверхности кистей грубеют и утолщаются.
Диагностические мероприятия
Диагностика порфирии выполняется гематологом. Врач осматривает ребенка или взрослого и включает в анамнез все симптомы, которые могут указывать на нарушения в процессе синтеза гема. Пациенту предстоит ответить на вопросы о принимаемых медикаментах, режиме питания, перенесенных инфекционных заболеваниях. Девушек врач расспрашивает о стабильности менструального цикла, беременностях и абортах.
Следующим этапом диагностики становятся лабораторные тесты. Пациентам назначаются:
- и биохимический анализы крови;
При наличии соответствующих показаний ребенок или взрослый посещает консультацию дерматолога, нефролога и гастроэнтеролога. Дифференциальная диагностика позволяет врачам исключить из анамнеза пациента неврологические и психиатрические патологии.
Терапевтический курс
Лечение острой и эритропоэтической порфирии требует госпитализации пациента. Лица, страдающие ПКП, могут проходить терапию в амбулаторных условиях. Современная медицина не располагает средствами, позволяющими полностью устранить нарушения в обмене порфиринов. Усилия врачей направлены на устранение симптомов заболевания и факторов, провоцирующих его обострения.
При острых формах патологии ребенку или взрослому назначаются производные аденозинтрифосфата, подавляющие синтез токсичного порфобилиногена. Дальнейшее лечение предусматривает прием пациентом больших дох глюкозы и переход на высокоуглеводную диету.
Эритропоэтический тип заболевания почти не поддается лечению. Основной терапевтической мерой становится ограничение пребывания пациента под солнечными лучами. Эрозии следует обрабатывать антисептическими средствами для предупреждения инфекционных поражений кожных покровов. Выраженный гемолиз (разрешение эритроцитов) становится показанием к удалению селезенки. В отдельных случаях пациентам показана трансплантация костного мозга.
При поздней кожной порфирии мужчинам и женщинам назначается плазмаферез и медикаментозная терапия. Применяемые препараты призваны снизить уровень всасывания порфиринов в ЖКТ.
Прогноз
Порфириновая болезнь относится к патологиям с неблагоприятным прогнозом. Пациенты с эритропоэтическим типом расстройства проживают 25–30 лет после постановки диагноза и начала лечения. Причиной смерти в 85% случаев становятся вторичные инфекции, развившиеся на фоне ослабления иммунитета человека. Острые порфирии завершаются летальным исходом в 15–20% клинически регистрируемых случаев. Смерть пациента наступает в результате паралича дыхательных мышц.
ПКП остается сравнительно благоприятной формой порфириновой болезни. Мужчинам и женщинам рекомендуется избегать провоцирующих осложнения факторов: скудного рациона, стрессов, избыточной инсоляции.
Диагностика и лечение порфирии в Москве
АО «Медицина» (клиника академика Ройтберга) обладает всем необходимым оборудованием для диагностики и лечения расстройств синтеза гема у детей и взрослых. Прием пациентов осуществляется в современном диагностическом комплексе, построенном с учетом последних достижений медицины.
Вопросы и ответы
Какой врач лечит порфириновую болезнь?
Лечение патологии осуществляется гематологом. При осложненном течении заболевания пациенту могут потребоваться консультации с другими врачами — иммунологом, терапевтом, нефрологом или гастроэнтерологом.
Передается ли порфирия от носителя к здоровому человеку?
Передача патологии от носителя к здоровому человеку невозможна. Заболевание наследуется детьми от родителей или приобретается под действием факторов риска.
Существует ли комплекс профилактических мер, позволяющих предупредить расстройство синтеза гема?
Современная медицина не располагает средствами для профилактики данной патологии. Носителям заболевания, планирующим рождение детей, следует посетить генетика. Врач изучит анамнезы потенциальных родителей и назначит паре необходимые анализы. Результаты лабораторных исследований позволят оценить вероятность развития порфириновой болезни у ребенка.
Поздняя кожная порфирия ( Урокопропорфирия , Хроническая печеночная порфирия )
Поздняя кожная порфирия – хроническое заболевание, характеризующееся повышенным образованием порфиринов и их накоплением в коже. При данной форме поражаются кожные покровы, подвергающиеся воздействию солнечного света (фотосенсибилизации). Патология проявляется гиперпигментацией участков кожи, повышенной ранимостью, гипертрихозом, образованием пузырей, эрозий и язв. Диагноз ставится на основании клинической картины, анамнеза, высокого содержания порфиринов в плазме, моче и кале. В качестве лечения проводятся сеансы плазмафереза и кровопусканий, назначаются аминохинолиновые и комплексообразующие препараты, солнцезащитные средства.
МКБ-10
Общие сведения
Поздняя кожная порфирия (ПКП, урокопропорфирия, хроническая печеночная порфирия) относится к группе заболеваний с нарушением порфиринового метаболизма (порфирий) и является самой распространенной из них. Средняя частота встречаемости урокопропорфирии составляет 1:10000 человек, а в странах Западной Европы и Южной Африки ‒ 1:1000 и 1:800 человек соответственно. Манифестация ПКП наступает в возрасте 40-50 лет. Болеют преимущественно мужчины, на долю которых приходится 90-93% всех клинических случаев. Данная патология характеризуется хроническим течением с рецидивами в весенне-летний период, что связано с увеличением длины дня и солнечной активности.
Причины ПКП
В основе развития заболевания лежит недостаточная активность уропорфириноген-декарбоксилазы, что ведет к накоплению порфиринов, в больших концентрациях оказывающих токсическое действие. Порфирины ‒ органические тетрапиррольные соединения, которые синтезируются главным образом в печени и костном мозге и необходимы для образования гемоглобина, миоглобина, каталаз, пероксидаз, цитохрома Р-450 и витамина В12. Сниженная функция уропорфириноген-декарбоксилазы приводит к торможению синтеза порфиринов на уровне уропорфирина и копропорфирина.
Это может произойти вследствие двух механизмов. Первый – мутация гена А1В18, кодирующего фермент, второй – действие различных факторов, оказывающих ингибирующее действие на фермент. К таким факторам относятся гепатит С, алкоголизм, хроническая интоксикация тяжелыми металлами (мышьяк, свинец, ртуть), прием пероральных контрацептивов, длительный контакт с нефтепродуктами. Наиболее частая причина ПКП у мужчин – алкоголизм и гепатит С, у женщин – гормональные контрацептивы. Более редкими причинами являются ВИЧ-инфекция, саркоидоз, злокачественная опухоль печени и терминальная стадия хронической почечной недостаточности. Наличие наследственной и приобретенной форм – главная отличительная особенность ПКП от других нарушений порфиринового обмена, которые являются только наследственными патологиями.
Патогенез
В результате сниженной функции уропорфириноген-декарбоксилазы увеличивается концентрация копропорфирина и уропорфирина. Данные вещества, накапливаясь в кожных покровах, под действием длинноволнового спектра солнечных лучей индуцируют образование свободных радикалов, что запускает процессы перекисного окисления липидов и высвобождение протеолитических ферментов из тучных клеток. Это приводит к клеточному повреждению базальной мембраны эпидермиса и сосудов дермы, что обусловливает отслойку наружного слоя кожи и появление симптомов фотосенсибилизации (легкая ранимость, пузыри, эрозии, язвы).
Отложение порфиринов в коже, а также стимуляция образования меланина меланоцитами способствует усилению пигментации. Гиперпигментация и фототоксические реакции также связаны с высоким содержанием железа в крови. Нарушение метаболизма железа является частой сопутствующей патологией ПКП, так как развивается вследствие хронических заболеваний печени. Механизм ускоренного роста волос на лице (гипертрихоза) до сих пор неизвестен. Поражение нервной системы ограничивается дисфункцией ее вегетативного отдела и нейротрофическими нарушениями. При гистопатологическом исследовании кожи больных ПКП обнаруживается ее субэпидермальная отслойка, разрыхление рогового слоя эпидермиса, отделение эпидермиса от соединительнотканной части, характерная фестончатость сосочков дермы и утолщение эндотелия поверхностных кожных сосудов.
Классификация
В зависимости от преобладания тех или иных клинических проявлений различают кожную, кожно-нервную, кожно-висцеральную и смешанную формы. По степени выраженности симптомов разделяют манифестную и латентную ПКП. По этиологическому фактору выделяют следующие виды урокопропорфирии:
Симптомы ПКП
Основной орган поражения – кожа. Наиболее типичными проявлениями манифестной формы считаются гиперпигментация, образование пузырей, гипертрихоз. Усиление пигментации наблюдается на участках кожи, систематически подвергающихся инсоляции (лицо, уши, шея, верхняя часть груди, кисти рук). Кожные покровы приобретают различные оттенки – от землисто-серого до бронзового. Интенсивность пигментации более выражена у брюнетов. В начале заболевания изменение цвета кожи носит временный характер, исчезающий в зимний период. По мере прогрессирования ПКП гиперпигментация становится постоянной. У некоторых пациентов долгое время это может быть единственным симптомом.
К признакам фоточувствительности относится повышенная ранимость кожи. Отслойка эпидермиса происходит даже при незначительных механических воздействиях. На коже образуются плотные пузыри размером до 1 см с жидкостным содержимым, которое может иметь серозный, гнойный или геморрагический характер. Через некоторое время пузыри вскрываются, оставляя после себя болезненные эрозии. После заживления эрозий формируются рубцы.
Для латентной ПКП характерны гипертрихоз височно-скуловой области лица, утолщение кожи на задней поверхности шеи с глубокими ромбовидными складками. Больные выглядят старше своих лет из-за появления или усиления морщин на лице. Вегетативно-трофические нарушения включают повышенную потливость, головные боли, нарушения сна, диарею или запор, учащение сердцебиения, усиление сосудистого рисунка на коже верхней части груди за счет пареза поверхностных капилляров (симптом «зарева» или «лимонной кожи»), деформацию ногтей, атрофию мышц кистей рук, предплечий, плечевого пояса.
Существуют атипичные варианты кожной порфирии. Язвенно-некротическая форма развивается у людей с ослабленным иммунитетом (сахарный диабет, ВИЧ-инфекция). При данном виде ПКП возникают глубокие язвенные дефекты кожи и гнойно-некротический распад мягких тканей. Инфильтративно-бляшечная, склеродермоподобная и витилигинозная формы имитируют изменения кожи при дискоидной красной волчанке (чешуйчатая эритема на лице в виде бабочки), диффузной склеродермии (уплотнение, кальциноз кожи) и витилиго (депигментированные пятна), что становится причиной частых диагностических ошибок.
Кожная порфирия у женщин имеет некоторые особенности течения: более раннее начало заболевания (около 30 лет), ограниченная пигментация на лице (хлоазмы), выраженный гипертрихоз, поражение нетипичных участков кожи – спина, бедра, подошвенная поверхность стоп. Среди женщин чаще, чем среди мужчин, встречаются витилигинозная, склеродермоподобная и инфильтративно-бляшечная формы ПКП.
Осложнения
Поздняя кожная порфирия ухудшает течение сердечно-сосудистых заболеваний, увеличивает риск развития сахарного диабета. Постоянные эрозии и язвенные дефекты кожных покровов часто осложняются бактериальной инфекцией. В редких случаях при язвенно-некротической форме развивается синдром системной воспалительной реакции и сепсис. Основную опасность для жизни представляют заболевания, которые вызывают ПКП: вирусный гепатит С и алкогольный гепатит, которые могут привести к циррозу и печеночной недостаточности, онкологические заболевания, хроническая почечная недостаточность, ВИЧ-инфекция.
Диагностика
Поздняя кожная порфирия представляет собой междисциплинарную проблему. В ее диагностике принимают участие врачи разных специальностей - дерматологи, гематологи, инфекционисты, гепатологи, ревматологи, онкологи. Так как в большинстве случаев кожная порфирия носит приобретенный характер, необходимо выяснить, на фоне какого заболевания она развилась. У пациента необходимо узнать об употреблении алкоголя, приеме оральных контрацептивов, наличии вирусного гепатита С. Для постановки диагноза проводятся следующие методы исследования:
Кожную порфирию следует дифференцировать с другими видами порфирий, протекающими с поражением кожных покровов (наследственная копропорфирия, врожденная эритропоэтическая и вариегатная порфирии). Также ПКП дифференцируют с дерматологическими (фотодерматозы, вульгарная пузырчатка, герпетиформный дерматит Дюринга), ревматологическими заболеваниями (дискоидная красная волчанка, диффузная склеродермия), гемохроматозом, надпочечниковой недостаточностью.
Лечение ПКП
Особое внимание при лечении ПКП уделяется устранению факторов и патологий, которые привели к развитию заболевания. Это подразумевает полный отказ от алкоголя, прекращение приема оральных контрацептивов, лечение гепатита С, проведение антиретровирусной терапии, химиотерапии и хирургического удаления злокачественной опухоли печени.
Патогенетическое лечение ПКП включает в себя несколько мероприятий. С целью удаления избытка порфиринов и железа из крови проводятся сеансы флеботомий (кровопусканий) и плазмафереза. В дополнение к этим процедурам, а также при противопоказаниях к ним используют лекарственные средства, связывающие и выводящие порфирины и железо. К ним относятся синтетические противомалярийные препараты из группы аминохинолинов (хлорохин, гидроксихлорохин) и комплексообразующие соединения (дефероксамин, Д-пеницилламин). При применении аминохинолинов очень важно начинать их прием с малых доз. Это связано с тем, что обычные дозы в начале лечения вызывают специфическую токсическую реакцию (порфириновый криз), характеризующуюся усиленным синтезом порфиринов и резким ухудшением состояния пациента (повышение температуры тела, тошнота, рвота, боли в животе). Для уменьшения абсорбции порфиринов в желудочно-кишечном тракте назначаются энтеросорбенты (активированный уголь).
Для защиты кожных покровов от солнечного света рекомендуется ношение максимально закрытой одежды, использование солнцезащитных кремов и мазей, содержащих парсол- 1789 или мексомил ‒ вещества, не пропускающие ультрафиолетовые лучи длинноволнового спектра. Обязательна постоянная обработка эрозивных поверхностей кожи антисептическими растворами во избежание инфекционных осложнений.
Прогноз и профилактика
Среди всех патологий с нарушением обмена порфиринов кожная порфирия является наиболее благоприятной в плане течения и прогноза. Серьезные осложнения возникают редко, их причиной в основном являются заболевания или патологии, которые приводят к развитию ПКП. Профилактика заключается в избегании факторов, способствующих возникновению кожной порфирии (употребление алкоголя, прием оральных контрацептивов, длительное пребывание на солнце, контакт с тяжелыми металлами, нефтепродуктами). Людям, у кого в семье есть больной наследственной формой ПКП, показано определение активности уропорфириноген-декарбоксилазы и проведение молекулярно-генетической диагностики.
2. Дерматовенерология. Национальное руководство/ под ред. Ю.С. Бутова, Ю.К. Скрипкина, О.Л. Иванова. — 2016.
3. Поздняя кожная порфирия/ Оркин В.Ф., Шабогина А.А., Олехнович Н.М., Кочнева Е.В.// Клиническая дерматология и венерология. 2015. № 3.
4. Поздняя кожная порфирия/ Монахов С.А.// Российский журнал кожных и венерических болезней – 2014 -№ 5.
Читайте также: