Профилактика дистрофинопатий. Миодистрофия Дюшенна.
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Тест позволяет выявлять мутации в гене дистрофина, которые ответственны за развитие наиболее частой причины поражения мышечной системы в молодом возрасте.
Синонимы русские
Дистрофинопатии, миодистрофия Дюшенна (МД), миодистрофия Беккера (МБ), ген DMD, генетическое обследование.
Синонимы английские
Dystrophinopathies, Duchenne muscular dystrophy, Becker muscular dystrophy, gene DMD.
Название гена
Локализация гена на хромосоме
Метод исследования
Полимеразная цепная реакция (ПЦР), фрагментный анализ гена DMD.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Миодистрофия Дюшенна/Беккера относится к X-сцепленным рецессивным наследственным заболеваниям и обусловлена мутацией в гене, кодирующем белок дистрофин (ген DMD). Ген дистрофина состоит из 79 экзонов – это один из самых крупных генов человека, расположен на Х-хромосоме (локус Xp21.2). При мышечной дистрофии обнаруживаются мутации (чаще всего делеции) одного или нескольких экзонов гена, реже точковые мутации или дупликации. В данном исследовании анализируются мутации экзонов 1-10, 21-30, 41-50, 61-70.
Дистрофинопатии представляют собой спектр наследственных Х-сцепленных заболеваний, вызываемых различными патологическими аберрациями в гене DMD. У носителей мутации мужского пола риск развития заболевания близок к 100%, у носителей женского пола проявления заболевания более мягкие либо не наблюдаются совсем. Тяжесть проявлений дистрофинопатий зависит от типов мутаций и может варьироваться от асимптоматического повышения креатинфосфокиназы или мышечных судорог с миоглобинурией до развития классических синдромов, таких как мышечная дистрофия Беккера и Дюшенна.
Миодистрофия Дюшенна (МД) чаще всего манифестирует в раннем детстве до 5 лет с задержки достижения основных этапов моторного развития и характеризуется полным отсутствием синтеза функционально активного дистрофина. На начальных этапах МД в основном поражаются проксимальные отделы мышечной системы (мышцы бедра, таза, плечевого пояса), но при последующей прогрессии затрагиваются все отделы мышечной системы. Помимо выраженной миодистрофии, у пациентов с МД наблюдается повышение уровня креатинфосфокиназы, псевдогипертрофия мышц голеней, различные скелетные аномалии и кардиомиопатия, чаще всего возникающая после 18 лет. Примерно у 75% пациентов с МД наблюдается делеция или дупликация одного или нескольких экзонов гена DMD.
Миодистрофия Беккера (МБ) представляет собой более легкую форму дистрофинопатии, характеризующуюся достаточным синтезом функционально активного белка. При МБ наблюдается мышечная слабость проксимальных отделов мышц, низкая толерантность к нагрузкам, миоглобулинурия, миалгия и повышение уровня креатинфосфокиназы. Делеции и дупликации одного или нескольких экзонов гена DMD являются наиболее частыми наблюдаемыми при МБ генетическими аберрациями (70-80% всех случаев).
Дистрофинопатия может проявляться у 5-10% носителей мутации женского пола мышечной слабостью, миалгией, судорогами и дилатационной кардиомиопатией.
Дистрофинопатии – X-сцепленные заболевания и наследуется по аутосомно-доминантному типу, то есть имеется 50% риска наследования данного заболевания от матери с аберрантным геном.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на миодистрофию Дюшенна/Беккера проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на миодистрофию Дюшенна/Беккера;
- при дифференциальной диагностике мышечной слабости;
- при дифференциальной диагностике мышечных судорог и миоглобинурии;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на выявлении делеции или дупликации одного или нескольких экзонов с помощью метода фрагментного анализа в гене DMD.
Патологических делеций и дупликаций экзонов 1-10, 21-30, 41-50, 61-70 в гене DMD не обнаружено.
Обнаружена делеция/дупликация в гене DMD. Диагноз "миодистрофия Дюшенна/Беккера" подтвержден.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Важные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
3 Общий анализ мочи с микроскопией
22 Креатинкиназа общая
47 Генетическое обследование на болезнь Кеннеди (спинальная и бульбарная мышечная атрофия) в гене AR
Прогрессирующая мышечная дистрофия Дюшенна — Беккера. Трудности диагностики
Цель статьи: представить клинический случай прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера и показать трудности и особенности диагностики этой генетической патологии.
Основные положения. В статье представлен анализ клинического случая диагностики ПМД Дюшенна — Беккера у мальчика 2 лет. Описаны этапы диагностического поиска, проведен анализ результатов клинического наблюдения и молекулярно-генетического обследования. Предложен алгоритм обследования при повышении активности аспартатаминотрасферазы (АСТ), аланинаминотрансферазы (АЛТ) неясного генеза.
Заключение. Описание клинического примера ПМД демонстрирует сложности, с которыми встречаются врачи разных специальностей при диагностике данного заболевания. Ранние клинические признаки, повышение в крови активности АСТ, АЛТ, креатинфосфокиназы являются основанием для назначения молекулярно-генетического исследования. Данную схему можно использовать при постановке диагноза ПМД, чтобы сократить длительные поиски несуществующей неврологической и инфекционной патологии, своевременно назначить адекватную терапию и улучшить качество жизни больного ребенка.
Вклад авторов: Царькова С.А. — проверка критически важного содержания, утверждение рукописи для публикации; Ушакова Р.А. — наблюдение, обследование пациента, сбор клинического материала, анализ и интерпретация данных; Громада Н.Е. — разработка концепции статьи, анализ и интерпретация данных, написание текста рукописи; Косенкова М.И., Мусалова О.Р. — обзор публикаций по теме статьи, написание текста рукописи.
Конфликт интересов: авторы заявляют об отсутствии возможных конфликтов интересов.
По данным Регистра врожденной и наследственной патологии Свердловской области клинико-диагностического центра «Охрана здоровья матери и ребенка», ежегодно выявляют 4–5 случаев прогрессирующей мышечной дистрофии (ПМД). Это труднокурабельное заболевание имеет высокую социальную значимость в связи с ранней инвалидизацией ребенка и необходимостью оказания своевременной психосоциальной помощи родителям и пациенту. Недооценка ранних симптомов ПМД Дюшенна — Беккера, клинических и нейрофизиологических критериев сопряжены с поздней диагностикой заболевания.
ПМД Дюшенна — Беккера — наследственное рецессивное нервно-мышечное заболевание, сцепленное с Х-хромосомой, вызванное мутациями в гене DMD , приводящими к отсутствию или недостаточной функции дистрофина, цитоскелетного белка, который обеспечивает прочность, стабильность и функциональность миофибрилл. Шифруется по классификации МКБ-10 как G71.0 2 .
Ген, отвечающий за выработку белка дистрофина, находится на Х-хромосоме (локализация Хр 21.2) и состоит из 79 частей-экзонов. При наличии мутаций в этом гене белок дистрофин не синтезируется, мышечная ткань гибнет, замещается жировой и соединительной тканью. В 40–60% случаев отмечается мутация (делеция — потеря или дупликация — удвоение) одного или нескольких экзонов [3, 4] .
Выделяют два клинических варианта: миодистрофии Дюшенна и Беккера. Миодистрофия Беккера (1 на 30 000 населения) — более легкий вариант заболевания, при котором синтез белка дистрофина идет не до конца, и в результате получается немного укороченный, но вполне функциональный белок. В данном случае болезнь протекает с медленным прогрессированием мышечной слабости и с сохранением способности к самостоятельной ходьбе в течение 15–20 лет от начала заболевания [3, 4] .
С миопатией Дюшенна рождается один из 5000 мальчиков (3,3 : 100 000 населения). Манифестация болезни чаще всего наблюдается в возрасте от 1 года до 5 лет. Заболевание характеризуется прогрессирующим злокачественным течением: формированием атрофии мышц тазового и плечевого пояса на фоне псевдогипертрофии икроножных, ягодичных, дельтовидных мышц, мышц живота и языка. Возможно снижение ментальной функции. Постепенно развивается деформация стоп, грудной клетки, позвоночника, прогрессируют дилатационная миокардиопатия и дыхательные нарушения, приводящие к летальному исходу в молодом возрасте [3, 4] .
Диагностические критерии заболевания следующие: пол пациента — мужской; установленный диагноз прогрессирующей миодистрофии у родственников мужского пола по материнской линии или неуточненное нервно-мышечное заболевание; установленный факт наличия в семье женщин-носительниц патологического гена; кардиологические заболевания (кардиомиопатии) у родственников женского пола по материнской линии; задержка становления двигательных навыков; снижение интеллекта; повышение активности трансаминаз АЛТ и АСТ и креатинфосфокиназы (КФК) в сыворотке крови.
Ранними симптомами заболевания являются повышенная утомляемость, мышечная слабость, гипотония в конечностях, псевдогипертрофия мышц голеней и бедер, частые падения или неуклюжесть, затруднение при приседании, беге, подъеме по лестнице, неспособность прыгать, использование вспомогательных приемов при подъеме с пола (ребенок помогает себе руками при подъеме из положения лежа, сидя), ходьба на носочках, задержка формирования речи [3, 4] .
В биохимическом анализе крови регистрируется повышение активности трансаминаз АЛТ, АСТ, лактатдегидрогеназы, КФК [5] .
При проведении электромиографии определяется миопатический тип с уменьшением (укорочением) величины средней длительности потенциала двигательных единиц (ПДЕ), снижением амплитуды отдельных ПДЕ.
На УЗИ мышц появляются признаки замены мышечной ткани жировой или фиброзной тканью. По данным ЭКГ, возможно появление аритмии, нарушение проводимости, на ЭхоКГ — признаки систолической дисфункции, дилатации левого желудочка, гипертрофии миокарда, митральной регургитации.
Для выявления дегенерации мышечной ткани используют МРТ и магнитно-резонансную спектроскопию мышц голени, бедер, таза (иногда денситометрию) [5] .
Генетические исследования MLPA (Multiplex Ligation-dependent Probe Amplification), секвенирование гена DMD предполагают проведение сравнительной геномной гибридизации массива и поиск точечных мутаций [6] .
В настоящее время лечение — симптоматическое, направленное на улучшение качества жизни. Применение ГКС позволяет замедлить прогрессирующую атрофию мышечной ткани. Препаратами выбора являются преднизолон или дефлазакорт. Назначение ингибиторов АПФ или β-блокаторов рекомендуют для профилактики дилатационной кардиомиопатии, мочегонных препаратов — при наличии сердечно-сосудистой недостаточности. Для профилактики остеопороза назначают препараты, содержащие витамин D2 и кальций [7] .
Детям с данной патологией рекомендуется физиотерапия, направленная на поддержание физической активности: профилактические растяжки, применение ортопедических аппаратов, лечебная физкультура [3] .
На стадии клинического эксперимента находится метод экзон-скиппинга — пропуска поврежденных экзонов, который предполагает «достройку» обходного параллельного пути для рамки считывания, минуя поврежденный экзон, что приводит к синтезу укороченного белка, сохраняющего свою функциональность.
Генно-клеточная терапия также находится в процессе разработки: использование стволовых клеток, «уснувшего» гена — утрофина (эмбрионального дистрофина), доставка в клетки генных конструкций микрогенов с помощью аденоассоциированных вирусов [8] .
Пациент находился под наблюдением в отделении патологии детей раннего возраста № 2 МАУ «Детская городская клиническая больница № 11» г. Екатеринбурга. У родителей ребенка получено добровольное информированное согласие на публикацию данных.
В результате ретроспективного анализа медицинской документации (индивидуальная карта развития ребенка Ф112/У) и опроса родителей выявлено, что мальчик рожден женщиной 22 лет от 1-й физиологически протекавшей беременности, в результате самопроизвольных срочных родов в головном предлежании плода на 39 неделе гестации. Масса тела ребенка при рождении — 3260 г, длина тела — 53 см, окружность головы — 36 см, окружность груди — 34 см, что соответствовало сроку гестации. Оценка по шкале Апгар — 8/9 баллов. Интранатальный и неонатальный периоды — без особенностей. Результат неонатального скрининга на наличие врожденных заболеваний отрицательный.
Наследственный анамнез не отягощен. Выписан из роддома на 3-и сутки домой. Вакцинация проведена согласно национальному календарю. Находился на естественном вскармливании до 9 месяцев.
Ребенок в течение первого года жизни рос и развивался соответственно возрасту: начал держать голову в 1 месяц, следить за игрушкой в горизонтальной плоскости, гулить — с 2 месяцев, брать предметы в руки — в 3,5–4 месяца, поворачиваться со спины на живот — в 5,5 месяцев, сидеть без поддержки — в 6 месяцев, ползать — с 9 месяцев, произносить слово «мама», стоять с опорой — в 12 месяцев, ходить самостоятельно — с 14 месяцев.
В возрасте 18 месяцев у ребенка выявлена гепатоспленомегалия (печень выступала из-под края реберной дуги на 3,5 см, селезенка — на 2 см), по данным УЗИ органов брюшной полости: правая доля печени — 74,5 мм, левая доля — 38 мм, длина селезенки — 78 мм, толщина — 34,2 мм. На фоне отсутствия синдрома желтухи и при нормативных показателях билирубина в анализе крови обнаружено стойкое повышение (в течение 6 месяцев) активности трансаминаз: АСТ — 360–390–426 Е/л, АЛТ — 298–417 Е/л (при норме до 45 Е/л и 40 Е/л соответственно), γ-глютамилтранспептидазы — 32,4 Е/л (при норме 6–23 Е/л). Пациент проконсультирован врачом иммунологом-инфекционистом. Методы ИФА и ПЦР-диагностики позволили исключить гепатит инфекционной этиологии.
На основании сведений, полученных от родителей, установлено, что после 16 месяцев жизни у мальчика появились скованность в ногах по утрам и неустойчивость при ходьбе, частые падения.
К 24 месяцам жизни физическое развитие по уровню биологической зрелости соответствовало паспортному возрасту. Масса — 14,5 кг. Рост — 93 см. Окружность головы — 48 см. Телосложение пропорциональное. Кожа эластичная, чистая. Сознание ясное. Очаговых и менингеальных симптомов нет. Чувствительная сфера и черепные нервы — без патологии.
Снижен мышечный тонус в нижних конечностях. Биципитальные и карпорадиальные, коленные и ахилловы рефлексы снижены и симметричны. Мышечная сила в верхних и нижних конечностях снижена до 4 баллов (определение по принципу «напряжения — преодоления»). Псевдогипертрофия икроножных мышц. Походка неуверенная. Позитивный симптом Говерса: поднимается из положения лежа и на корточках, опираясь руками о пол и колени. Сидит и опирается на руку. Бег затруднен. Не подпрыгивает. Частично понимает речь, выполняет инструкции матери. Произносит отдельные слова.
Функции слуха и зрения сохранены. Функции тазовых органов не нарушены.
Используя скрининговые центильные графики подуровней нервно-психического развития для индивидуальной оценки тестируемых навыков и умений [9], мы определили, что подуровень ручной умелости, развития речи и социальной адаптации находится в пределах 25–75% центильной зоны и соответствует возрасту. Подуровень общей моторики по тестируемым навыкам и поздние сроки их появления находятся за пределами 90% центильной зоны и свидетельствуют о задержке и дисгармоничном развитии.
Со стороны органов дыхания и сердечно-сосудистой системы отклонений нет. Данные ЭКГ и ЭхоКГ — без патологии. Результаты электронейромиографии: аксональная нейропатия малоберцового нерва слева.
С учетом описанного выше неврологического дефицита, пола пациента, признаков синдрома цитолиза, стойкого увеличения активности трансаминаз на фоне отсутствия желтушного синдрома решено провести исследование уровня КФК в крови. Выявлено превышение показателя в 70 раз — 17 453 Е/л (при референсных значениях до 247 Е/л). Это стало показанием для поиска заболеваний, ассоциированных с наследственными формами ПМД.
Проведено молекулярное-генетическое исследование. На первом этапе делеции и дупликации в гене DMD методом MLPA не обнаружены. Затем с помощью молекулярно-генетического метода массового параллельного секвенирования найден вариант в гемизиготном состоянии в 13 экзоне гена DMD (chrX:32613972 G>A, c.1504C>T) (точечная мутация). Описан ранее в литературе как патогенный. Валидирован методом прямого секвенирования по Сэнгеру.
Тип наследования — X-сцепленный рецессивный. Генетический риск для сибсов: по заболеванию для братьев — до 50%, по носительству для сестер — 50%. Для уточнения риска необходимо проведение молекулярно-генетической диагностики у матери для поиска выявленной мутации в гене DMD .
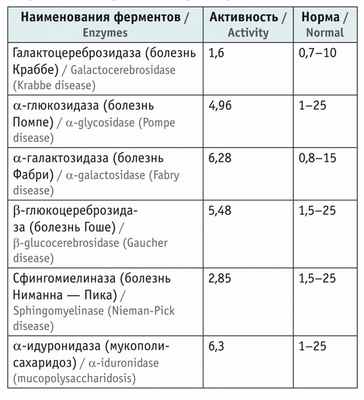
Активность лизосомных ферментов в крови у пациента находилась в пределах референсных значений ( табл. ).
Показатели активности лизосомных ферментов в пятнах высушенной крови, мкМ/л/ч
Ребенку назначено лечение: преднизолон в дозе 0,5 мг/кг/сутки. Рекомендована физиотерапия с дозированной физической нагрузкой. Предложена социально-психологическая помощь родителям: работа с психологом, даны координаты Благотворительного фонда помощи детям с миодистрофией Дюшенна и иными тяжелыми нервно-мышечными заболеваниями «МойМио».
Из представленных данных следует, что ребенку в возрасте 2 лет на основании жалоб, данных анамнеза, особенностей неврологического статуса, результатов лабораторно-инструментальных и молекулярно-генетических исследований был поставлен диагноз: мышечная дистрофия Дюшенна — Беккера, стадия сохраненной способности к самостоятельному перемещению. Проведение энзимодиагностики позволило исключить некоторые наследственные лизосомные болезни накопления (см . табл. ).
У пациента дебют заболевания наступил в возрасте 2 лет. Минимальные клинические признаки ПМД остались незамеченными, родители первыми обратили внимание на нарастание мышечной слабости после 16–18 месяцев, что явилось поводом обращения к педиатру и неврологу. Симптомы дистрофии прогрессировали медленно с неравномерным поражением мышц, в дебюте заболевания пострадали отдельные группы мышц, что привело к относительной компенсации двигательных расстройств.
По нашим и литературным данным, в структуре ошибочного диагноза доминируют перинатальная энцефалопатия, гепатит, гепатоз, кардиомиопатия, суставно-мышечная патология [3, 4] . У наблюдаемого ребенка отмечались стойкое длительное (в течение 6 месяцев) увеличение активности трансаминаз, наличие гепатоспленомегалии, что заставило исключить гепатит инфекционной этиологии. Высокие показатели КФК обнаружили позже.
В данном случае, при миодистрофии Дюшенна, повышение активности АЛТ и АСТ в анализах крови имеет внепеченочное происхождение и определяется при разрушении миофибрилл мышц на фоне высоких показателей КФК.
Известно, что алгоритм диагностики ПМД состоит из нескольких этапов. Если ребенок начинает ходить поздно, выявлен положительный симптом Говерса или другие нарушения мышечной функции, имеется отягощенный семейный анамнез, в крови — увеличение активности трансаминаз неуточненного генеза, то рекомендуется определение уровня КФК. В свою очередь, высокие показатели КФК создают необходимость проведения молекулярно-генетической диагностики мутации гена дистрофина и биопсии мышц ребенка [3] .
Для структурирования диагностического поиска заболевания мы использовали адаптированный алгоритм Р.А. Ушаковой [10] . Согласно представленной схеме, у пациента наблюдалось повышение активности трансаминаз неясного генеза, положительный симптом Говерса, отсутствие ПМД в семейном анамнезе ( рис. ).
Рис. Алгоритм диагностики прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера [10]
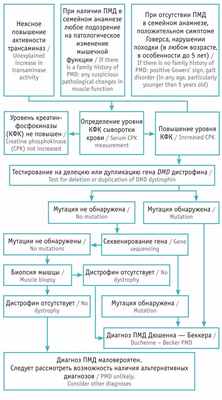
Для верификации диагноза прогрессирующей мышечной дистрофии (ПМД) Дюшенна — Беккера при наличии у пациента клинической симптоматики, характерной для данного заболевания, необходимо проведение молекулярно-генетического исследования с целью поиска мутации в гене DMD . Аналогичное обследование предлагают матери пациента для определения типа наследования, расчета риска патологии при планировании следующей беременности.
Данную схему можно использовать при постановке диагноза ПМД даже при отсутствии пренатальной диагностики, указаний на заболевание в семейном анамнезе, чтобы сократить длительные поиски несуществующей неврологической и инфекционной патологии, обратить внимание на ранние признаки заболевания, своевременно провести определение уровня креатинфосфокиназы, молекулярно-генетическое исследование, назначить адекватную терапию и улучшить качество жизни больного ребенка.
Профилактика дистрофинопатий. Миодистрофия Дюшенна.
Профилактика дистрофинопатий. Миодистрофия Дюшенна.
Профилактика дистрофинопатий (Дюшенна и Беккера) заключается в широком внедрении методов пренатальной диагностики в амбулаторную практику, выявлении носителей патологического гена, развитие сети генетического консультирования. Необходимо проведение анализа родословных больных и носителей, определение уровня креатинкиназы в крови, клиническое, гистологическое, электромирграфическое обследование, биопсия мышц для выявления состояния дистрофина, ПЦР-диагностика для выявления гетерозиготного носительства. Аномалии плода могут быть диагностированы внутриутробно с помощью молекулярного исследования ворсин хориона на ранних сроках беременности (после 8 недель) или методом амниоцентеза.
После 19 недель гестации под контролем ультразвука можно получить образцы мышцы плода для выявления дистрофина. Если женщина является носителем патологического гена, то шанс родить здорового ребенка составляет около 50%.
В остальных случаях рождается больной миодистрофией Дюшенна мальчик или девочка-носительница патологического гена. Однако приблизительно в 30% случаев семейный анамнез больных миодистрофией Дюшенна не отягощен. Для больных миотонической миодистрофией очень важна профилактика серьезных сердечно-сосудистых осложнений перед проведением хирургического вмешательства. Необходимо определение уровня инсулина в крови, исследование в свете щелевой лампы, ЭКГ и адекватное лечение. С. Общие принципы лечения 1. Миодистрофия Дюшенна
Образовательные программы для больных и членов их семей оказывают значительную поддержку пациентам и их родственникам. Люди подвергаются не только огромным стрессовым нагрузкам, но и сталкиваются со значительными финансовыми сложностями при уходе и.лечении больных миодистрофией Дюшенна. Основной задачей членов семьи является продление периода самостоятельного, а затем колясочного передвижения больного ребенка, вовлечение его в общественную жизнь. Если нет отставания умственного развития, то мальчик должен посещать нормальную школу. Ребенок нуждается в специальных приспособлениях для компенсации недостатка движения и слабости верхних конечностей.
ЛФК и физиотерапия используется для поддержания подвижности и профилактики развития ранних контрактур в сгибателях бедра, в коленных и голеностопных суставах. Хотя не найдено способа предотвратить прогрессирование заболевания, можно добиться продления периода подвижности больного при помощи пассивных нагрузок на мышцы и суставы и адекватной ортопедической поддержки. Упражнения также полезны для профилактики развития ожирения, которое, в свою очередь, затрудняет движения ребенка и мешает нормальной работе органов дыхания. В тот период, когда больные теряют способность к ходьбе, им необходимо стоять, по крайней мере, в течение трех часов в день (за несколько приемов). Следует избегать постельного режима, который усугубляет слабость. Однако интенсивные физические нагрузки противопоказаны.

Ноги у ребенка с мышечной дистрофией Дюшенна
При невозможности передвигаться без посторонней помощи требуется обучение пользованию костылями. Возможно применение хирургических методов лечения. При помощи шин можно удерживать суставы больного в нейтральном положении. Шины надевают на ночь. Когда больной постоянно находится в кресле-коляске, значительно усиливается сколиоз и нарастают контрактуры. Хирургическое лечение сколиоза обычно не применяют из-за риска развития осложнений при общей анестезии. Методикой выбора является техника сегментарной спинальной стабилизации Luque. Однако хирургическое вмешательство по поводу сколиоза становится необходимым, если он приводит к затрудненности дыхания, при том условии, что угол сколиоза менее 40 градусов. У больных миодистрофией Дюшенна очень большой риск развития опасных осложнений при проведении общей анестезии. Нельзя применять сукцинилхолин и галотан, т. к. нередко наблюдается синдром, напоминающий проявления злокачественной гипертермии.
Для снижения выраженности нежелательных эффектов применяют недеполяризующие миорелаксанты.
Дыхательная терапия. Качество жизни больных можно улучшить регулярным проведением дыхательной гимнастики или игрой на духовых музыкальных инструментах. На поздних стадиях заболевания следует периодически назначать механическую вентиляцию легких, особенно тем больным, у которых отмечается гиперкапния в крови, в связи с чем нарушается ночной сон. Адекватные дыхательные упражнения служат лучшей профилактикой присоединения легочной инфекции.
Лекарственные препараты. Преднизолон в дозе 0,75 мг/кг/сут назначают только в отдельных случаях, например, при обострении заболевания. Преднизолон обычно увеличивает мышечную силу через месяц после начала лечения. Своего максимума положительный эффект достигает примерно на четвертый месяц лечения. Однако применение преднизолона ограничено из-за выраженных побочных эффектов. К ним относятся бессонница, поведенческие расстройства, нарушения со стороны желудочно-кишечного тракта. Контрольные исследования по применению преднизолона показали, что длительность его положительных эффектов составляет не более трех лет. При использовании преднизолона больной должен соблюдать диету с ограничением поваренной соли и жиров. При контакте с больными ветряной оспой ребенку с мышечной дистрофией назначают профилактические препараты специфического (противоветряночного) иммуноглобулина.
Генная терапия проходит стадию ранних клинических испытаний. Испытания трансплантации миобластов, проведенные на мышах mdx, продемонстрировали хорошие результаты, однако это лечение оказалось неэффективным у людей. Генная терапия с использованием вирусных векторов или липосом находится на стадии доклинических испытаний (в моделях на животных).
Принципы лечения больных миодистрофией Беккера не отличаются от лечения миодистрофии Дюшенна. В легких случаях требуется только внимательное врачебное наблюдение и профилактические мероприятия.
Миодистрофия Эмери-Дрейфуса требует тщательного контроля функции сердца с целью своевременной имплантации водителя ритма для предотвращения угрожающих жизни аритмий.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Адекватный менеджмент пациентов с дистрофинопатиями (мышечная дистрофия Дюшенна/Беккера): применение объективизирующих шкал и дополнительных методов исследования
В отечественной медицинской практике отсутствует четкая стандартизация подходов к курации больных миодистрофией Дюшенна/Беккера (МДД/МДБ). Это зачастую приводит к снижению качества жизни пациентов и, что наиболее важно, уменьшению ее продолжительности. В статье отражен накопленный за последнее десятилетие опыт западных коллег, в том числе объединенный в соглашении консенсуса 2010 г. по курации пациентов с МДД, дополненный наблюдениями из собственной клинической практики. Описаны стандартные шкалы оценки степени ограничения моторных навыков и мышечной слабости у пациентов с МДД/МДБ, а также представлен алгоритм мультидисциплинарного подхода с фокусом внимания на профилактике, своевременной диагностике и лечении основных осложнений заболевания и терапии кортикостероидами: кардиоваскулярных, ортопедических, респираторных и т.д. Применение представленных рекомендаций позволяет не только значительно улучшить качество и продолжительность жизни пациентов с МДД/МДБ, но и участвовать в мультицентровых исследованиях с целью поиска патогномоничного и симптоматического лечения.
Ключевые слова
Об авторах
Региональная общественная организация «Общество специалистов по нервно-мышечным болезням», Москва
Россия
Список литературы
1. Emery A.E. The muscular dystrophies. Lancet 2002;359:687.
2. Rivier F., Meyer P., Walther-Louvie U. et al. Врожденные мышечные дистрофии: классификация и диагностика. Нервно-мышечные болезни 2014;1;6–20.
3. Bushby K., Connor E. Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig (Lond)2011;1(9):1217–35.
4. Mayhew A.G., Cano S.J., Scott E. et al. Detecting meaningful change using the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Dev Med Child Neurology 2013;55:1046–52.
5. Montes J., Gordon A.M., Pandya S. et al. Clinical outcome measures in spinal muscular atrophy. J Child Neurol 2009;24(8):968–78.
6. Kissel J.T., Scott C.B., Reyna S.P. et al. SMA carni-VAL trial part II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One 2011;6(7):e21296.
7. Swoboda K.J., Scott C.B., Crawford T.O. et al. SMA Carni-VAL trial part I: doubleblind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 2010;5(8):e 12140.
8. Bushby K., Finkel R., Birnkrant D.J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177.
9. Darras B.T. Treatment of Duchenne and Becker muscular dystrophy. Official Topic from UpToDate®. 2014. 23 p.
10. Nolan M.A., Jones O.D., Pedersen RL., Johnston H.M. Cardiac assessment in childhood carriers of Duchenne and Becker muscular dystrophies. Neuromuscul Disord 2003;13:129.
11. Colan S.D. Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation 2005;112:2756.
12. Duboc D., Meune C., Lerebours G. et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005;45:855.
13. Jefferies J.L., Eidem B.W., Belmont J.W. et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005;112:2799.
14. Passamano L., Taglia A., Palladino A. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myologica 2012; XXXI: 121–5.
15. Finder J.D., Birnkrant D., Carl J.et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med 2004;170:456.
16. Birnkrant D.J., Bushby K.M., Amin R.S. et al. The respiratory management of patients with duchenne muscular dystrophy: a DMD care considerations working group specialty article. Pediatr Pulmonol 2010;45:739.
17. Do T. Orthopedic management of the muscular dystrophies. Curr Opin Pediatr 2002; 14:50.
18. Cheuk D.K., Wong V., Wraige E. et al. Surgery for scoliosis in Duchenne muscular dystrophy. Cochrane Database Syst Rev 2013; 2:CD005375.
19. Bachrach L.K. Taking steps towards reducing osteoporosis in Duchenne muscular dystrophy. Neuromuscul Disord 2005; 15:86.
20. Biggar W.D., Bachrach L.K., Henderson R.C. et al. Bone health in Duchenne muscular dystrophy: a workshop report from the meeting in Cincinnati, Ohio, July 8, 2004. Neuromuscul Disord 2005;15:80.
21. Quinlivan R., Roper H., Davie M. et al. Report of a Muscular Dystrophy Campaign funded workshop Birmingham, UK, January 16th 2004. Osteoporosis in Duchenne muscular dystrophy; its prevalence, treatment and prevention. Neuromuscul Disord 2005;15:72.
Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера
(Мышечная дистрофия Дюшенна; мышечная дистрофия Беккера)
, MDCM, New York Presbyterian Hospital-Cornell Medical Center
Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера являются Х-сцепленными рецессивными Сцепленные с Х-хромосомой рецессивные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения расстройствами, характеризующимися прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией мышечных волокон. Дистрофия Беккера имеет позднее начало и вызывает более легкие симптомы. Диагноз предполагают клинически и подтверждают генетическим исследованием или анализом белкового продукта (дистрофина) мутантного гена. Лечение направлено на поддержание функции при помощи физикальной терапии, использования ортопедических скоб и ортопедических аппаратов. Пациентам с миодистрофией Дюшенна необходимо лечение преднизоном или дефлазакортом, а иногда терапия путем пропуска экзонов с использованием антисмысловых олигонуклеотидов.
Мышечные дистрофии являются наследственными прогрессирующими заболеваниями мышечной системы, возникающими из-за дефектов в одном или нескольких генах, необходимых для нормальной структуры мышц и их функционирования. Дистрофические изменения (например, некроз и регенерация мышечных волокон) видны на биоптатах.
Дистрофия Дюшенна и дистрофия Беккера являются вторыми по распространенности мышечными дистрофиями (после плече-лопаточно-лицевой миопатии Плече-лопаточно-лицевая мышечная миопатия Лице-лопаточно-плечевая мышечная дистрофия является наиболее распространенным типом мышечной дистрофии. Большинство случаев манифестируют к 20 годам. Для этого заболевания характерна слабостью. Прочитайте дополнительные сведения ). Эти нарушения обусловлены мутациями гена дистрофина (dystrophin) – крупнейшего человеческого гена, известного науке, расположенного в локусе Xp21.2. Около 70% случаев дистрофии Дюшенна вызываются делецией или дупликацией одного или нескольких экзонов. При дистрофии Беккера 85% пациентов имеют делецию, а 10% имеют дупликацию.
При дистрофии Дюшенна эти мутации приводят к тяжелому отсутствию ( 5%) дистрофина, белка мембраны мышечных клеток. При дистрофии Беккера мутации приводят к образованию аномального дистрофина или его недостаточности.
Дистрофия Дюшенна и дистрофия Беккера в совокупности поражают приблизительно 1/5000–1/6000 живорожденных мальчиков; подавляющее большинство из них имеют дистрофию Дюшенна. У женщин-носителей может наблюдаться бессимптомное повышение уровня креатинкиназы и, в некоторых случаях, гипертрофия задней части голени.
Клинические проявления
Дистрофия Дюшенна
Это заболевание поражает около 10 из 100 000 живорожденных мальчиков и проявляется, как правило, в возрасте 2–3 лет. Слабость затрагивает проксимальные мышцы, как правило, сперва нижних конечностей. Дети часто ходят на пальцах, имеют походку вразвалку и лордоз. Таким детям сложно бегать, прыгать, подниматься по ступенькам и вставать с пола. Они часто падают и получают переломы рук или ног (примерно у 20% больных). Наблюдается стабильное прогрессирование слабости, и почти у всех детей развиваются сгибательные контрактуры конечностей и сколиоз Идиопатический сколиоз Идиопатический сколиоз – боковое искривление позвоночника. Диагноз устанавливают на основании клинических признаков, в том числе по результатам рентгенографии брюшной полости. Методы лечения. Прочитайте дополнительные сведения Последствия вовлечения сердечной мышцы включают в себя дилатационную кардиомиопатию Дилатационная кардиомиопатия Дилатационная кардиомиопатия – нарушение функционирования миокарда, приводящее к сердечной недостаточности, при которой преобладают дилатация желудочков и систолическая дисфункция. Симптомы. Прочитайте дополнительные сведения и аритмии Аритмии Введение (Overview of Arrhythmias) Здоровое сердце бьется регулярным, скоординированным образом благодаря тому, что электрические импульсы в сердце генерируются и распространяются миоцитами с уникальными электрическими свойствами. Прочитайте дополнительные сведенияДистрофия Беккера
По сравнению с дистрофией Дюшенна дистрофия Беккера поражает
Диагностика
Анализ ДНК на наличие мутаций
Иногда, мышечная биопсия с иммунным окрашиванием дистрофина
Диагноз подозревают в зависимости от характерных клинических признаков, возраста начала заболевания и семейного анамнеза, предполагающего Х-связанный рецессивный тип наследования Сцепленные с Х-хромосомой рецессивные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения . Миопатические изменения видны на электромиографии (потенциалы моторных единиц быстро возрастают, имеют небольшую продолжительность и низкую амплитуду) и, при её выполнении, биопсия мышц показывает некроз и заметное изменение размера мышечных волокон, не отделенных от моторных единиц. Уровни креатинкиназы превышены в 100 раз по сравнению с нормой.
Мутационный анализ ДНК лейкоцитов периферической крови с использованием мультиплексной лигазно-зависимой амплификации ДНК-зондов (MLPA) является основным подтверждающим тестом; он может выявить аномалии в гене dystrophin. Если патология не выявляется при мультиплексной лигазно-зависимой амплификации ДНК-зондов (MLPA), но все еще подозревается дистрофия Дюшенна или Беккера, могут проводить полное секвенирование гена dystrophin для обнаружения небольших генетических изменений, например, точечных мутаций.
Если при генетическом тестировании диагноз не подтверждается, следует провести анализ дистрофина с иммунным окрашиванием биоптатов мышц. Дистрофин у пациентов с дистрофией Дюшенна не обнаруживается. У пациентов с дистрофией Беккера дистрофин, как правило, ненормальный (низкий молекулярный вес) или присутствует в низкой концентрации.
Пациенты с дистрофией Дюшенна должны подвергаться оценке исходного состояния сердечной функции при помощи ЭКГ и ЭхоКГ на момент постановки диагноза или до 6-летнего возраста.
Выявление носительства и пренатальная диагностика возможны с помощью обычных исследований (например, изучения родословной, определения креатинкиназы, пола плода) в сочетании с анализом рекомбинантной ДНК и иммуноокрашиванием дистрофина в мышечной ткани.
Лечение
Иногда корректирующая хирургия
Иногда при кардиомиопатии используют ингибиторы ангиотензин-превращающего фермента и/или бета-блокаторы
При дистрофии Дюшенна – преднизон или дефлазакорт, иногда – антисмысловые олигонуклеотиды (методы терапии на основе пропуска экзонов)
Не существует никакого специфического лечения. Легкие (т.е. субмаксимальные) активные упражнения рекомендуется выполнять как можно дольше во избежание дисфункциональной атрофии или осложнений от гиподинамии. Пассивные упражнения могут продлить период способности к передвижению. Ортопедические вмешательства должны быть направлены на поддержание функции и предотвращение контрактур. Ортез голеностопного сустава, одетый на время сна, может помочь предотвратить сгибательные контрактуры. Ортопедические аппараты на ногах могут временно помочь сохранить способность стоять и передвигаться. Иногда необходима корректирующая хирургия, в частности при сколиозе. Следует избегать ожирения; потребности в калориях, как правило, будут ниже, чем обычно, из-за снижения физической активности.
Дыхательную недостаточность иногда можно лечить с помощью применения неинвазивной респираторной поддержки (например, через назальную маску— Астматический статус Астматический статус ). Элективная трахеотомия получает все большее признание, что позволяет детям с дистрофией Дюшенна доживать до возраста старше 20 лет.
Детям с дилатационной кардиомиопатией Дилатационная кардиомиопатия Дилатационная кардиомиопатия – нарушение функционирования миокарда, приводящее к сердечной недостаточности, при которой преобладают дилатация желудочков и систолическая дисфункция. Симптомы. Прочитайте дополнительные сведенияЭкспериментальные методы лечения при дистрофии Дюшенна и дистрофии Беккера включают генную терапию, креатин, инактивацию миостатина, миогенные клетки-предшественники и антиоксидант идебенон.
Варианты лекарственной терапии при дистрофии Дюшенна
Согласно самым последним рекомендациям, при дистрофии Дюшенна у пациентов > 5 лет, у которых выявлена задержка или регресс моторных навыков, настоятельно рекомендован ежедневный прием преднизона или дефлазакорта ( 1 Справочные материалы по лечению Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера являются Х-сцепленными рецессивными расстройствами, характеризующимися прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией. Прочитайте дополнительные сведения ). Эти препараты начинают работать через 10 дней после начала терапии; пик эффективности приходится на 3-й месяц и сохраняется в течение 6-ти месяцев. Длительное применение улучшает силу, отодвигает возраст, при котором возникает потеря способности передвигаться, на 1,4-2,5 года, улучшает временные функциональные тесты (измерение быстроты выполнения функциональной задачи ребенком, например, ходьбы или вставания с пола), улучшает легочную функцию, уменьшает ортопедические осложнения (например, необходимость хирургического лечения сколиоза), стабилизирует сердечную функцию (например, задерживает начало кардиомиопатии до 18 лет) и увеличивает выживаемость от 5 до15 лет. ( 1 Справочные материалы по лечению Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера являются Х-сцепленными рецессивными расстройствами, характеризующимися прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией. Прочитайте дополнительные сведения ). Назначение преднизона через день неэффективно. Увеличение веса и кушингоидное лицо являются самыми распространенными побочными эффектами во временном интервале от 6 до 18 месяцев после начала приема. Риск компрессионного перелома позвоночника и переломов длинных костей также увеличивается. Дефлазакорт может быть связан с большим риском развития катаракты, чем преднизон. Использование преднизона или дефлазакорта при дистрофии Беккера еще не были должным образом изучены.
Для лечения миодистрофии Дюшенна была одобрена терапия путем пропуска экзонов. К такому методу лечения относится применение трех препаратов: этеплирсена, голодирсена и вилтоларсена. Эти препараты называются антисмысловыми олигонуклеотидами и действуют как молекулярные участки аномального гена dystrophin, в котором отсутствует один или более экзонов (отсутствующие экзоны препятствуют сборке полного белка, вызывая тем самым тяжелые симптомы). Препараты маскируют экзон таким образом, что он будет пропущен и игнорирован во время продукции белка, что позволяет продуцировать белок дистрофин, который, хотя и является ненормальным, но способен выполнять свои функции и может уменьшить симптомы, так что они становятся более похожи на таковые у мальчиков с менее тяжелым течением мышечной дистрофии Беккера.
Этеплирсен пропускает экзон 51. Ограниченные данные свидетельствуют о том, что прием этеплирсена приводит к повышению уровня дистрофина в мышцах и повышению эффективности ходьбы при проведении тестов на время у 13% пациентов с дистрофией Дюшенна, имеющие dystrophin генную мутацию, которая может пропускать экзон 51. Одобрение препарата подверглось критике, потому что оно было основано на небольшом исследовании, которое основывалось на суррогатном исходе (дистрофин при мышечной биопсии), а клиническая польза осталась недоказанной. Рекомендуемая доза этеплирсена составляет 30 мг/кг внутривенно в течение 35–60 минут 1 раз в неделю.
Голодирсен и вилтоларсен пропускают экзон 53. Их можно использовать у 8% пациентов с дистрофией Дюшенна, имеющих мутацию гена дистрофина, которую можно корректировать путем пропуска экзона 53. Клиническая польза остается недоказанной. Рекомендуемая доза голодирсена составляет 30 мг/кг внутривенно в течение 35–60 минут 1 раз в неделю, а доза вилтоларсена – 80 мг/кг внутривенно 1 раз в неделю.
Аталурен (PTC124) - препарат для перорального применения, доступный в Европейском союзе и Великобритании для лечения генетических дефектов, вызванных нонсенс-мутациями (стоп-кодон). Это вариант для пациентов с дистрофией Дюшенна в возрасте 2 лет и старше, которые лечатся амбулаторно и чье заболевание вызвано бессмысленными мутациями, которые являются причиной слишком раннего прекращения продукции белка дистрофина в клетке, что приводит к появлению неспособного нормально функционировать белка. Клиническая польза препарата также не доказана, и в США он пока еще не одобрен к использованию ( 2 Справочные материалы по лечению Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера являются Х-сцепленными рецессивными расстройствами, характеризующимися прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией. Прочитайте дополнительные сведения ).
Справочные материалы по лечению
1. Gloss D, Moxley RT 3rd, Ashwal S, Oskoui M: Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 86:465–472, 2016. doi: 10.1212/WNL.0000000000002337
2. McDonald CM, Campbell C, Torricelli RE, et al: Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:(10101):1489–1498, 2017. doi: 10.1016/S0140-6736(17)31611-2
Ключевые моменты
Дистрофия Дюшенна и дистрофия Беккера являются рецессивными расстройствами, связанными с Х-хромосомой, которые вызывают снижение концентрации дистрофина – белка в мембранах мышечных клеток.
Пациенты имеют значительную, прогрессирующую слабость, что вызывает тяжелую форму физической несостоятельности, в том числе трудности при ходьбе, частые падения, дилатационную кардиомиопатию и раннюю смерть вследствие дыхательной недостаточности.
Пациентам полезно делать активные и пассивные упражнения наряду с применением ортопедических аппаратов для ног и ортезов голеностопного сустава.
У больных с дистрофией Дюшенна ежедневный прием преднизона или дефлазакора повышает мышечную силу и массу, улучшает функцию внешнего дыхания и помогает отстрочить развитие кардиомиопатии, однако часто провоцирует побочные эффекты.
Пациентам, страдающим дистрофией Дюшенна с определенными мутациями, можно также назначить этеплирсен, голодирсен или вилтоларсен, несмотря на ограниченные данные об их клинической пользе.
Применение ингибитора ангиотензин-превращающего фермента и/или бета-блокатора может помочь предотвратить или замедлить развитие кардиомиопатии.
Вспомогательная вентиляция легких (неинвазивная, а позже и инвазивная) может способствовать продлению жизни.
Дополнительная информация
Ниже следуют некоторые англоязычные ресурсы, которые могут быть информативными. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов.
Muscular Dystrophy Association: Информация об исследованиях, лечении, технологиях и поддержке для пациентов, живущих с Duchenne muscular dystrophy и Becker muscular dystrophy
National Organization for Rare Disorders: Подробная информация относительно Duchenne muscular dystrophy и Becker muscular dystrophy, в том числе стандартное и экспериментальное лечение и ссылки на смежные темы
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: