Прогерия
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Прогерия, или синдром Хатчинсона – Гилфорда — редкое заболевание из группы ламинопатий, характеризующееся преждевременным старением с поражением кожи, костей и сердечно-сосудистой системы. В основе патогенеза лежат патогенные варианты в гене LMNA, приводящие к аномалии морфологии ядерной мембраны, нарушению экспрессии генов, изменению структуры хроматина, дисфункции митохондрий, дефектам репарации ДНК и альтернативного сплайсинга, ускорению укорочения теломер. Основные проявления болезни — поражения кожи (склеродермоподобный синдром и нарушение пигментации), липодистрофия, позднее прорезывание и скученность зубов, алопеция, дистрофия ногтей, явления остеолизиса концевых фаланг, вальгусная деформация тазобедренных суставов и контрактуры суставов, атеросклероз, тугоухость, ранние инфаркты и инсульты. Склеродермоподобные изменения кожи, остеопороз, сгибательные контрактуры межфаланговых суставов кистей, остеоартрит тазобедренных суставов требуют дифференциальной диагностики с ревматическими заболеваниями. Основой ведения больных с прогерией являются профилактика и лечение сердечно-сосудистых проявлений заболевания (ранних инсультов и инфарктов, артериальной гипертензии и атеросклероза), повышение качества жизни и ежедневной активности пациентов. Изучается эффективность терапии больных с прогерией с применением ингибиторов фарнезилтрансферазы (монотерапия; комбинация с бисфосфонатами или статинами), ретиноидов и 1,25(OH)2 — витамина D3. Настоящий литературный обзор дополнеНописанием клинического случая прогерии у девочки. Диагноз подтвержден секвенированием гена LMNA (по Сэнгеру), выявлен ранее описанный патогенный вариант гена в экзоне 11 (c.1824C>T, rs58596362) в гетерозиготном состоянии (p.Gly608Gly, NM_170707.3).
Ключевые слова
Об авторах
Бучинская Наталья Валерьевна, кандидат медицинских наук, педиатр, ревматолог, врач-генетик консультативного отделения.
Автор подтвердил отсутствие конфликта интересов, о котором необходимо сообщить.
Автор подтвердил отсутствие конфликта интересов, о котором необходимо сообщить.
Автор подтвердил отсутствие конфликта интересов, о котором необходимо сообщить.
Санкт-Петербургский государственный педиатрический медицинский университет; Национальный медицинский исследовательский центр им. В.А. Алмазова
Россия
Автор подтвердил отсутствие конфликта интересов, о котором необходимо сообщить.
Список литературы
3. Hutchinson J. Case of congenital absence of hair, with atrophic condition of the skin and its appendages, in a boy whose motherhad been almost wholly bald from alopecia areata from the age of six. Lancet. 1886;1:923.
5. Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford progeria syndrome. 2003 Dec 12 [Updated 2019 Jan 17]. In: GeneReviews [Internet]. Adam MP, Ardinger HH, Pafon RA, et al., eds. Seattle (WA): University of Washington, Seattle; 1993–2020.
21. Huang S, Liang Y, Wu W, et al. Analysis of a case with typical Hutchinson-Gilford progeria syndrome with scleroderma-like skin changes and review of literature. Zhonghua Er Ke Za Zhi. 2014;52(2):112–116.
Рецензия
При поддержке: Подготовка материалов рукописи выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации (Соглашение № 075-15-2022-301 от 20.04.2022)
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Аннотация
Напечатать статью
Посмотреть метаданные
Как ссылаться
Поиск ссылок
Послать статью по эл. почте (Необходимо имя пользователя (логин))
Связаться с автором (Необходимо имя пользователя (логин))
Н. В. Бучинская
Диагностический центр (медико-генетический)
Россия
Бучинская Наталья Валерьевна, кандидат медицинских наук, педиатр, ревматолог, врач-генетик консультативного отделения.
А. Ж. Ахенбекова
Казахский национальный медицинский университет им. С Д. Асфендиярова
Казахстан
А. А. Бугыбай
Научный центр педиатрии и детской хирургии
Казахстан
М. М. Костик
Санкт-Петербургский государственный педиатрический медицинский университет; Национальный медицинский исследовательский центр им. В.А. Алмазова
Россия
В Британии умерла 17-летняя девушка: ее тело было как у 104-летней
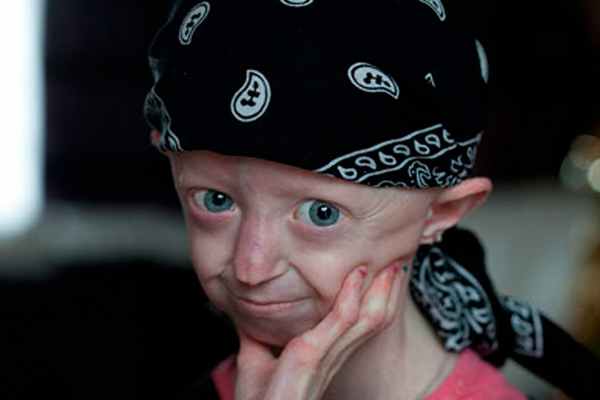
Безжалостная смерть
Несмотря на смертельно опасное заболевание — синдром Хатчинсона — Гилфорда, — девушка дожила до 17 лет. Хотя медики утверждали, что жить ей от силы 13 лет и ни годом больше. Хейли Окинс умерла на родине, хотя незадолго до этого проходила курс лечения в США, где принимала инновационные препараты, которые замедляют процесс старения. Однако затем девушка заболела пневмонией и была вынуждена вернуться в Великобританию.
«Моя девочка ушла в лучший из миров. Последний вдох она сделала в 21.39», — написала в своем фейсбуке ее мама.
Не остались безучастными к трагедии и представители общества по исследованию синдрома Хатчинсона — Гилфорда, написавшие, что «скорбят вместе с семьей Хейли Окинс — умной, красивой и вдохновлявшей многих английской розы».

«Она ушла от нас, но не из наших воспоминаний. Ушла из зоны нашей доступности, но не из наших сердец», — резюмировали в обществе.
При жизни Хейли Окинс стала настоящей медийной звездой и активно привлекала внимание общественности к борьбе с неизлечимым синдромом Хатчинсона — Гилфорда: про нее сняли документальные фильмы «Девушка, которая старше своей матери» и «Самый старый тинейджер: необычные люди». Кроме того, в 14 лет она выпустила автобиографию, озаглавленную «Постаревшая раньше времени». При жизни она успела посетить многие страны, встретиться со многими знаменитостями — например, с принцем Чарльзом и Джастином Бибером.
Ее мать вспоминала, что при рождении Хейли была обычным светловолосым и голубоглазым ребенком. И лишь, когда ей стукнуло 13 месяцев, а она продолжала носить одежду для младенцев, они пошли к врачу и узнали страшную правду о неизлечимой болезни.
Молодые мои старики
Название «прогерия» произошло от древнегреческих слов προσ — «сверх», γέρων – «старик». Прогерия — это редкое заболевание, известное также как прогерический синдром Хатчинсона — Гилфорда, которое вызывается спорадической мутацией всего лишь одного гена. Спорадическими называют мутации, которые не передаются ребенку по наследству от родителей, а возникают при созревании половых клеток или при развитии эмбриона.
Важно понимать, что прогерия — болезнь генетическая, но не наследственная.
Виновный ген Imna кодирует белок под названием «преламин А», из которого формируется оболочка клеточного ядра. Преламин А, созданный по лекалам гена-мутанта, делает эту оболочку несовершенной, что приводит к изменению структуры ядра, нестабильности генома и нарушению экспрессии генов.
В результате дети, больные прогерией, быстро лысеют, у них стареет кожа, становятся хрупкими кости.
В шесть лет они выглядят стариками, а примерно в тринадцать умирают от инсульта или инфаркта из-за сильного атеросклероза.
Этим, собственно, и объясняется спорадический характер мутации: больные детской прогерией просто не успевают дожить до репродуктивного возраста и передать дефект по наследству.
Впервые болезнь была описана в 1886 году известным английским врачом Джонатаном Хатчинсоном и независимо от него в 1897 году его соотечественником Гастингсом Гилфордом.
Лечения пока нет, но оно поможет и здоровым
Прогерия — это редкое заболевание. По данным медиков из Нидерландов, прогерия встречается в одном случае на четыре миллиона. В настоящий момент в мире живут около 100 людей с этим заболеванием.
Несколько людей смогли пережить «критический» 13-летний возраст и дожили до 26 лет, как, например, южноафриканец Леон Бота, который успел сделать себе творческую карьеру — он известен как художник, музыкант и диджей.

Леон Бота — южноафриканский художник, музыкант и диджей. Также известен как один из немногих доживших до 26-летнего возраста людей, больных прогерией
Максимум, что можно сделать для больного, это постоянно предпринимать точечные профилактические меры, чтобы этим продлить жизнь. Тот же Леон Бота в 20-летнем возрасте перенес операцию на сердце, которая должна была предотвратить сердечные приступы из-за атеросклероза, который является причиной смерти более 90% людей, страдающих прогерией. Это позволило прожить ему еще пять с лишним лет.
Правда, положительные результаты были получены в экспериментах на мышах, и до лечения людей дело за прошедшие два года, согласно списку публикаций группы профессора Бергё, пока не дошло.
Между тем ученые весьма заинтересованы в том, чтобы полностью разобраться в вопросе лечения прогерии.
«Причина этого интереса очевидна: это необычайное сходство между ходом нормального процесса старения и ускоренного старения у больных прогерией детей, а это значит, что прогерия может прояснить многие тайны и нормального старения, — считает профессор Бергё. — У детей развивается остеопороз, мышечная слабость, инфаркт миокарда, инсульт — все как у старых людей, но у них никогда не бывает ни рака, ни слабоумия».
Прогерия Хатчинсона-Гилфорда LMNA м.
Ген LMNA А/С (LAMIN A/C) кодирует белок ламин. Находится на хромосоме 1 в регионе 1q22. Состоит из 12 экзонов.
Мутации в гене ламина приводят также к развитию таких заболеваний, как дилятационная кардиомиопатия с нарушением ритма и проводимости (ДКМП), тип 1А, мышечная дистрофия Эмери-Дрейфуса 2, аутосомно-доминантная, мышечная дистрофия Эмери-Дрейфуса 3, аутосомно-рецессивная, синдром рука-сердца словенского типа, наследственная моторно-сенсорная нейропатия тип 2 B1, семейная частичная липодистрофия, синдром Малуф, врожденная мышечная дистрофия, мышечная дистрофия поясноконечностная тип 1В, мандибулоакральная дисплазия, летальная рестриктивная дермопатия.
Наличие мутаций в гене ламинов А/С в гетерозиготном состоянии обнаружено также у нескольких больных с клиническими проявлениями синдрома Вернера, однако, в большинстве случаев синдром Вернера наследуется по аутосомно-рецессивному типу и обусловлен мутациями в гене RECQL2.
Прогерия — редкое наследственное заболевание, проявляющееся преждевременным и ускоренным старением организма.
Ген LMNA кодирует преламин А, который является предшественником зрелых форм ламинов А и С. Все мутации в гене ламинов А/С, приводящие к возникновению этого синдрома, нарушают процесс сплайсинга в области 3` конца гена LMNA, что приводит к нарушению процессинга преламина А и его накоплению в клетке. Ряд авторов предлагают называть этот мутантный преламин «прогерином». В клетках больных прогерией ядерные оболочки сморщиваются, ядра приобретают неправильную форму. Такие клетки не могут нормально делиться. В результате организм не только перестает расти, но и теряет способность заменять отмирающие клетки новыми, что и приводит к ускоренному старению.
Возрастом начала прогерии Хатчинсона-Гилфорда считают 1-2 года, но возможен и более ранний дебют. Больные имеют характерный внешний вид: низкий рост, относительно большую голову и уменьшенную лицевую часть черепа, небольшой тонкий клювовидный нос («птичье» лицо), микрогнатию, экзофтальм, тотальную алопецию, оттопыренные уши. Частыми симптомами являются: алопеция (врожденная или начинается до полутора лет, сопровождается разрушением волосяных фолликулов); истонченная с первых месяцев жизни кожа, выбухающие вены на волосистой части головы; врожденные или возникающие в грудном возрасте склеродермоподобные уплотнения внизу живота, на бедрах, ягодицах; очаговая гиперпигментация открытых участков тела, усиливающаяся с годами. Гипоплазия ногтей. Ногти ломкие, желтоваты, утолщенные, выпуклые (симптом часовых стекол). Подкожная жировая клетчатка атрофируется с раннего детства, дольше сохраняется на щеках и лобке (вес больных обычно не превышает 20кг). С 1-2 лет развивается фиброз околосуставных тканей; контрактура локтевых и коленных суставов придает больному «позу всадника». Характерны гипоплазия, дисплазия и дегенеративные изменения скелета; позднее прорезывание молочных и постоянных зубов; скученность зубов; адентия и гиподонтия; множественный кариес. Уже с пяти лет у пораженных детей развивается распространенный атеросклероз, особенно аорты, коронарных и брыжеечных артерий; в более позднем возрасте могут появиться шумы в сердце и гипертрофия левого желудочка.
Ранний атеросклероз сокращает продолжительность жизни, основная причина смерти – инфаркт миокарда. Известен случай ишемического инсульта.
По уровню умственного развития дети с прогерией Хатчинсона-Гилфорда не уступают сверстникам, а часто и опережают их.
Прогерия
Прогерия – редкий синдром ускоренного старения, который проявляется в раннем детстве и вызывает преждевременную смерть.
Прогерия вызывается спорадической мутацией LMNA-гена, который кодирует белок (ламин А), обеспечивающий молекулярную поддержку клеточных ядер. Дефект белка приводит к нестабильности ядра при делении клеток и ранней гибели всех клеток тела.
Симптомы и признаки прогерии развиваются в течение 2 лет и включают:
Задержку роста (например, низкий рост, позднее прорезывание зубов)
Черепно-лицевые аномалии (например, черепно-лицевая диспропорция, микрогнатия, крючковатый нос, макроцефалия, большой родничок)
Физические признаки, связанные со старением (например, морщинистая кожа, облысение, снижение амплитуды движений суставов, жесткая кожа, напоминающая склеродермию)
Диагностика прогерии обычно очевидна по внешнему виду, однако проводиться дифференциальная диагностика с сегментарной прогерией (например, акрогерией, метагерией) и другими причинами недостаточности роста. Средний возраст наступления смерти – 12 лет; причина – заболевания коронарных артерий и сосудов головного мозга. Могут развиться резистентность к инсулину и атеросклероз. Следует отметить, что другие проблемы, связанные с нормальным старением (например, увеличение риска развития рака, дегенеративный артрит), отсутствуют.
Прогерия на сегодняшний день не поддается лечению. Доступны группы поддержки.
Другие прогероидные синдромы
Преждевременное старение является особенностью других редких прогероидных синдромов.
Синдром Вернера – это преждевременное старение после полового созревания с истончением волос и развитием состояний, характерных для старости (например, катаракты, сахарного диабета, остеопороза, атеросклероза). Синдром Ротмунда-Томсона проявляется преждевременным старением с повышенной восприимчивостью к раку. Оба вызваны генной мутацией, приводящей к дефекту ДНК-геликазы RecQ, которая в норме участвует в репарации ДНК.
Синдром Коккейна является аутосомно-рецессивным расстройством, вызванным мутацией в гене ERCC8, который играет важную роль в эксцизионной репарации ДНК. Клинические симптомы включают серьезное нарушение роста, кахексию, ретинопатию, гипертонию, почечную недостаточность, светочувствительность кожи и умственную отсталость.
Неонатальный прогероидный синдром (синдром Видемана–Раутенштрауха) является рецессивным наследуемым синдромом старения, приводящим к смерти до достижения 2 лет.
Другие синдромы (например, Дауна Синдром Дауна (трисомия 21) Синдром Дауна является аномалией 21-й хромосомы, может проявляться нарушением умственного развития, микроцефалией, небольшим ростом и характерным внешним видом. Диагноз предполагают на основании. Прочитайте дополнительные сведения ) иногда имеют черты прогерии.
Дополнительная информация
Ниже следуют некоторые англоязычные ресурсы поддержки, которые могут стать полезными родителям и лицам, осуществляющим уход. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов:
Progeria Research Foundation: Ресурс, посвященный поиску метода излечивания и эффективного лечения прогерии, который на своей веб-сайте предоставляет информацию, ресурсы и поддержку
Share and Care Cockayne Syndrome Network: Ресурс, предоставляюший группу поддержки семьям и специалистам, интересующихся синдромом Коккейна
Rothmund-Thomson Syndrome Foundation: Ресурс, который предоставляет образовательные материалы касательно синдрома Ротмунда-Томсона и связанных с ним состояний и поддерживает клинические исследования
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Синдром Гетчинсона-Гилфорда (прогерия)
Синдром Гетчинсона-Гилфорда или прогерия (сенильный нанизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения. Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом. Этиология прогерии неясна. Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у больных. Однако исследования зарубежных ученых позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения. Характерен вид лица: с экзофтальмом, тонким клювовидным носом, большим мозговым и малым лицевым черепом, голос тонкий, имеются скелетные аномалии. Пубертат обычно не наступает, наружные гениталии гипоплазированы. Интеллект средний или выше среднего. Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Ключевые слова
Для цитирования:
For citation:
Синдром Гетчинсона-Гилфорда (Hutchinson-Gilford Progeria Syndrome), или прогерия (сенильный папизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения.
Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом.
Этнология прогерии неясна.
Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у бальных. Однако исследования зарубежных ученых [2-4, 7] позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Отмечается влияние возраста отца как возможной причины новых мутаций. Так, средний возраст отцов составляет 35-37 лет.
Аутосомно-рецессивный тип наследования синдрома также обсуждается в литературе [7, 8, И]. Этот тип наследования был впервые предположен Gabr и соавт. в 1960 г. при описании двух моиозиготных сестер, а впервые сообщил о семейном случае прогерии Paterson в 1922 г., хотя описание двух больных братьев было неполным и без фотографий.
Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения.
Дети рождаются нормальными, но к 1-му году жизни наблюдается выраженная задержка роста и массы тела. Конечный рост в среднем достигает 100 см. В первые годы жизни развивается тотальная алопеция. Кожа топкая, лоснящаяся, сухая, тугонатянутая (па кистях и стопах, наоборот, морщинистая). В нижней части живота и бедрах кожные изменения напоминают склеродермию. С возрастом появляются коричневые пигментные пятна. Потовые и сальные железы атрофируются.
Подкожный жировой слой полностью отсутствует, за исключением лобковой области. На черепе выражена подкожная венозная сеть. Нощи дистрофичные, влоть до аплазии. Зубы прорезываются с задержкой, аномально расположены, с ранним разрушением как молочных, так и постоянных зубов.
Характерен вид лица: с экзофтальмом, гонким клювовидным носом, большим мозговым и малым лицевым черепом. Голос тонкий.
Скелетные аномалии включают резорбцию ключицы с замещением фиброзной тканью, резорбцию конечных фаланг кистей (акроостеолиз), истончение длинных трубчатых костей и ребер, тугоподвижность сутавов пальцев, увеличение локтевых и коленных суставов. Часты асептические некрозы головки бедренной кости и вывих тазобедренного сустава.
Пубертат обычно не наступает, наружные гениталии ги- поплазированы.
Интеллект средний или выше среднего.
Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Следует отметить, что заболевания, характерные для нормального процесса старения (катаракта, опухоли, сахарный диабет), встречаются при прогерии крайне редко.
Прогноз для жизни неблагоприятный: продолжительность жизни колеблется от 7 до 28 лет, в среднем составляя 12-13,5 года [1, 4, 5, 7, 10]. Основные причины летальных исходов - острый инфаркт миокарда, застойная сердечная недостаточность, инсульты. На аутопсии выявляются распространенный атеросклероз, гипоплазия гонад, иногда гипоплазия надпочечников и значительная гиперплазия тимуса, истончение коркового слоя костей.
Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Гормональные исследования у детей [4] выявляют нормальную ночную секрецию соматотропного гормона гипофиза, но крайне низкий уровень инсулиноподобного фактора роста (ИФР-1) в плазме крови. Это позволяет предполагать наличие у данных больных бионеактивпого пула СТГ в крови, либо периферическую резистентность к эндогенному СТГ. либо выраженный дефицит питания. Трехмесячный период высококалорийного питания не увеличивает уровень ИФР-1 в крови, но ускоряет линейный рост.
Прогерия считается состоянием, связанным с ннсулиноре- зистентностыо умеренной степени. В 1983 г.[9] впервые была описана девочка, у которой в 2-легнсм возрасте уровень инсулина в крови натощак составлял 20-40 мкг/дл. В 4 года через 3 мес после удаления кисты яичника развилась гипергликемия натощак (до 250 мкг/дл) с высоким уровнем инсулина (более 2200 мкЕД/мл) и С-пептида (32,4 нг/мл) в крови. Уровень гемоглобина А1 достигал 10%. Связывание инсулина с рецепторами эритроциов было в пределах нормальных значений.
Иммунологический аспект в патогенезе прогерии впервые был выдвинут Walford в 1970 г. В 1973-1976 г. Singal и Goldstein выявили отсутствие либо резкое снижение экспрессии HLA культурой фибробластов кожи у детей с прогерией. Вместе с тем другие исследователи, анализируя экспрессию HLA у детей с прогерией и их здоровых родственников [2], нс выявили ни количественного, ни качественного дефицита в экспрессии HLA в фибробластах кожи. Различие в частоте встречаемости ряда HLA-антнгенов у больных прогерией и здоровых людей не дает право в настоящее время говорить о специфической ассоциации HLA и прогерии в связи с малым числом исследуемого материала.
Биохимическими исследованиями показано, что одним из биомаркеров старения является мочевая экскреция гиалуроновой кислоты. В норме у детей и подростков содержание ее составляет менее 1% от уровня общих гликозаминогликанов и увеличивается с возрастом до 5-6%. У детей с прогерией выявлено значительное повышение (до 10-20%) экскреции гиалуроновой кислоты с мочой по сравнению со здоровыми людьми [4]. Данное повышение не наблюдается ни при одном генетическом заболевании, кроме синдрома Вернера, или “прогерии взрослых” [4]. Считается, что гиалуроновая кислота является ключевым фактором антиангиогенеза в процессе созревания и старения.
Изучение культуры фибробластов кожи от пациентов с прогерией выявляет значительное снижение клеточного роста вследствие подавления митотической активности. С другой стороны, отмечается нормальное распределение типов коллагена в коже, характерное для детей, а именно, преобладание коллагена 3-го типа над коллагеном 1-го типа [8].
Имеются данные, что в основе прогерии лежит дефицит метаболизма витамина Е [7].
Ряд исследователей связывают прогерию с генетически обусловленной ошибкой в синтезе внутриклеточных белков. Так, показано, что эритроциты больных детей содержат повышенную термолабильную фракцию ферментов: глюкозо-6- фосфатдегидрогепазу и б-фосфоглюконатдегидрогепазу |6]. Другие работы не подтверждают эту концепцию [3].
Данные олене или детей с прогерией крайне малочисленны.
Патогенетически оправданными считаются терапия витамином Е для восполнения его дефицита (Лугез и МШап, 15974) и усиленное белковое питание.
Приводим описание собственного клинического случая.
Боль пая Н., 3 лет 9 мес., поступила в детское отделение ЭНЦ РАМН с жалобами на отставание в росте и массе, сниженный аппетит, облысение, резкую головную боль.
Раннее развитие: держит голову с 1 мес жизни, сидит с 6 .мес жизни, ходит с 1 года 2 мес, зубы появились в 1 год 1 мес, говорит с 1,5 лет. Грудное вскармливание - до 1 года 8 мес.
Перенесенные заболевания: дисплазия тазобедренный суставов (в 1 мес жизни), двусторонний врожденный вывих бедер (диагностирован в 6 мес жизни), стоматит, легкая форма (в 3 года).
Аллергологический анамнез не отягощен.
Наследственность по низкорослости не отягощена. Мать 24 лет, рост 165 см, родственники. со стороны матери: бабушка - рост 157 см, дедушка - 180 см, тетя - 168 см. Отец 26 лет, рост 176 см; родственники со стороны отца: бабушка - рост 157 см, дедушка - 170 см. Отягощена наследственность по сахарному диабету II типа, который имеется у бабушки со стороны отца и у прабабушки со стороны матери. Отягощена наследственность по бронхиальной астме, тяжелая форма которой имеется у прадедушки, со стороны матери и у двоюродной прабабушки со стороны матери.
Анамнез заболевания: с 2-месячного возраста замечены уплотнение кожных покровов и подкожной жировой клетчатки, лоснящаяся кожа на бедрах, животе, ягодицах, цианоз носогубного треугольника. В 3-месячном возрасте диагностирована легкая форма склеродермии. При исследовании биоптата кожи выявлен гиперкератоз эпителия и умеренный склероз дермы. Консультирована дерматологом: диагноз склеродермии был снят, больше данных, свидетельствующих о склеродерме. Получала лечение преднизолоновой мазыо в течение 3 мес с умеренным эффектом - блеск и плотность кожных покровов уменьшились. В возрасте 1 года появилась венозная сеть на голове. Отмечалась гипотрофия П-Ш степени. Невропатолог диагностировал перинатальную энцефалопатию, компенсированную гидроцефалию. В 1 год 7 мес девочка была впервые консультирована эндокринологом: диагностирована задержка физического развития смешанного генеза. Масса тела 7800 г, костный возраст соответствовал паспортному. В 1 год 10 мес начали выпадать полосы на голове. В 1 год 11 мес впервые консультирована генетиком, поставлен диагноз: “Синдром Гетчинсона—Гилфорда”. Девочка была обследована в стационаре по месту жительства: рост 73 см, масса 7900 г. На ЭКГ: метаболические изменения миокарда
Данные обследования в ЭНЦ РАМН. Хронологический возраст 3,9 года. Рост 81,2 см. Коэффициент стандартного отклонения (SDS роста) - 4,35. Масса тела 9,5 кг. Рост сидя 48,5 см, коэффициент “верхний ссгмент/пижпий сегмент" 1,48. Окружность головы 49 см.
Отмечаются ярко выраженные черты синдрома Гетчинсона - Гилфорда: 1) крупная голова с диспропорционально большим мозговым черепом и малым лицевым. Вдавлен) гость височных костей; 2) выраженная венозная сеть на голове; 3) тотальная алопеция; 4) узкий, деформированный нос с истончением кожи на нем, цианоз носогубного треугольника; 5) истончение кожи на туловище, конечностях, морщинистость кожи на ладонях и ступнях. Склеродермоподобные изменения кожи на животе, спине и ягодицах - очаги депигментации диаметром 0,5-0,7 см, множественные, уплотненные. Депигментация сосков; 6) подкожная жировая клетчатка развита слабо, распределена равномерно; 7) гипоплазия ногтей кистей и стоп; утолщение межфаланговых суставов и концевых фаланг кистей и стоп; варусная девиация верхней трети предплечий, короткая шея (см. рисунок).
Область сердца визуально не изменена. Тоны сердца ясные, ритмичные. Часота сердечных сокращений 104 удара в минуту, АД 80/50 мм рт.ст. Дыхание везикулярное, хрипов нет. Зев чистый. Живот мягкий, безболезненный. Печень не увеличена, селезенка не пальпируется. Стул регулярный. Дизурических расстройств нет. Симптом Пастернацкого отрицательный с обеих сторон. Эндокринный статус: щитовидная железа не увеличена, симптомов нарушения функции нет. Симптомы гипокортицизма отсутствуют. Половой статус препубертатный: Ах 0, Р 0, Ма 0, Me 0.
Гормональное исследование крови: кортизол 321,4 нмоль/л, общий трийодтиронин 2,48 нмоль/л, общий тироксин 100,0 нмоль/л, ТТГ 2,10 мкЕД/л, пролактин 248,0 мкЕД/л, ЛГ 1,60 ЕД/л, ФСГ 5,50 ЕД/о, 17-оксипрогестерон 1,5 нг/мл. Соматотропный гормон на фоне стимуляционной пробы с клофелином: 0 мин - 1,7 нг/мл, 30 мин - 1,5 нг/мл, 60 мин - 2,3 нг/мл, 120 мин - 71,0 нг/мл, 150 мин - 29,3 нг/мл.
Гормональное исследование мочи: свободный кортизол 1115,0 нмоль/л на 1 г креатинина (креатинин 0,24 г/л). Дс- гидроэпиандростерон-сульфат — следы.
Показатели гуморального аутоиммунитета: у больного ребенка не выявлено антител пи к тиреоглобулину человека, ни к микросомальному антигену тиреоцигов, ни к поверхностным антигенам клеток аденогипофиза и клеток коры надпочечников крысы.
Вместе с тем у матери ребенка обнаружены антитела к поверхностным антигенам клеток аденогипофиза крысы, а у бабушки по материнской линии - слабо положительная реакция па наличие антител к микросомальному антигену тиреоцигов при отсутствии антител к другим изучаемым антигенам.
Рентгенографическое исследование: на рентгенограмме черепа и кистей структура костей не изменена. Форма и размеры турецкого седла обычные. Сосудистый рисунок костей свода усилен. Дифференцирование скелета соответствует 12- 15 мес. Ногтевые фаланги деформированы, треугольной формы. На рентгенограмме стоп отмечается небольшое уплотнение стенок arteria dorsalis pedis.
Компьютерная томография головного мозга: на серии компьютерных томограмм изменения плотности мозговой ткани не выявлено. Желудочковая система не изменена. Хиазмальная цистерна расширена. Несколько расширена межполушарная щель. Данных, свидетельствующих об объемном процессе головного мозга, не выявлено.
ЭКГ: ЧСС 120 в минуту, ритм синусовый. Электроэнцефалограмма: на фоне умеренных диффузных изменений биоритмики с признаками диэнцефальной заинтересованности отмечаются признаки ирригации стволово-диэнцефальных структур с легким акцентом справа, в теменно-затылочной области. Эхоэнцефалограмма: эхо-пульсация неустойчиво усилена до 55%, смещения срединных структур нет, легкое расширение 111 желудочка (до 6 мм), вентрикулярный индекс умеренно выше нормы. Заключение: смещения срединных структур не выявлено. Расширение боковых желудочков.
Ультразвуковое исследование: щитовидная железа: типично расположена, контуры ровные, структура гомогенная. Правая доля 2,3x1,1хо,9 см, слева - 2,4x1,2x0,9 см, толщина перешейка 0,4 см. Объем щитовидной железы 2,55 мл. Надпочечники: нс увеличены. Органы малого таза: размеры матки и яичников соответствуют возрастной норме. Матка: 3,1x1,3x1,1 см, правый яичник: 1,6x1,2x0,8 см, левый яичник: 1,4x1,2x1,0 см. Печень: увеличена правая доля - 8,5 см, левая доля 2,6 см. Структура паренхимы гомогенная, внутри- печеночные протоки не расширены. Воротная вена не расширена. Желчный пузырь конкрементов не содержит, Почки: топография не изменена, размеры в пределах возрастной нормы. Структуры хорошо дифференцированы, без гидро- нефротических изменений и достоверных эхо-признаков дополнительных объемных включений. Паренхима гомогенна, толщина се соответствует возрастной норме.
Консультация окулиста: моторно-зрачковых нарушений не выявлено. Передний отрезок и среды без патологии. Глазное дно: диски бледно-розовые, сосуды умеренно расширены, полнокровны, извиты по всей протяженности сетчатки. Сетчатка на периферии разряжена, небольшие отеки. Заключение: повышение внутричерепного давления.
Консультация невропатолога: субкомпепсированпая гидроцефалия на фоне скудной церебральной симптоматики.
Консультация дерматолога: Alopecia totalis. Рекомендованы курсы лечения препаратами, улучшающими микроциркуляцию (трептал, троксевазип), поливитамины (Bj, В2, В(„ В15, А, Е); наружно — втирание с массажем головы геля актовегина, геля троксевазина, димексида, 01. Ricini.
Учитывая имеющиеся в литературе данные об эффективности применения гормона роста у детей с прогерией [4], для увеличения линейного роста девочке был назначен пробный 3-месячный курс лечения рекомбинантным гормоном роста человека “SAIZEN” (ARES-SERONO). Недельная доза составила 15 ЕД/м 2 , суточная доза — 1 ЕД, подкожно, ежедневно, 7 раз в неделю.
Лечение рекомендовано проводить под контролем гликемии и суточной глюкозурии.
Помимо гормона роста, девочке назначено лечение, рекомендованное дерматологом, и полноценное белковое питание.
Таким образом, представленный материал, основанный на данных анамнеза, жалоб, результатах клинического обследования и лабораторно-инструментальных исследований, подтверждает наличие у ребенка классического синдрома Гетчинсона-Гилфорда (прогерии).
Читайте также: