Рентгенограмма костей при гипофосфатазии
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Что такое гипофосфатазия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 7 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
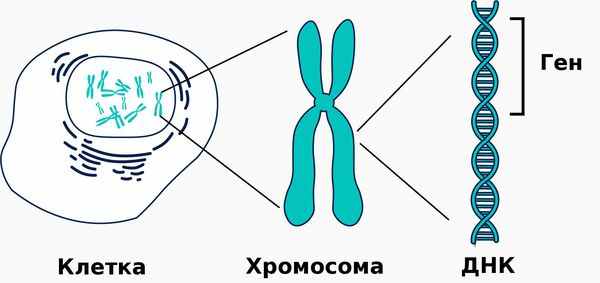
Гипофосфатазия — это наследственное метаболическое заболевание, связанное со снижением активности либо с полным отсутствием тканенеспецифической щелочной фосфатазы. Эта редкая болезнь затрагивает многие органы и системы и является жизнеугрожающей для пациента. [20] [23]
Такой фермент, как щелочная фосфатаза, играет важную роль в минерализации тканей. Поэтому её дефицит становится причиной различных системных нарушений, проявляющихся недостаточной минерализацией костей и их деформацией. При тяжёлых формах гипофосфатазии у новорождённых также развивается дыхательная недостаточность и судороги.
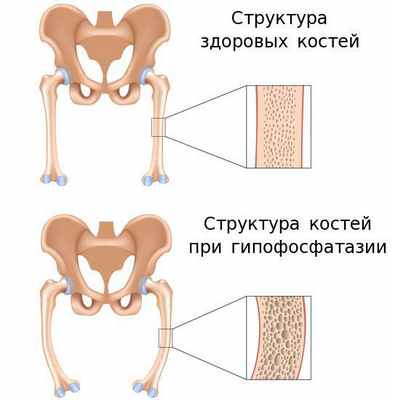
Причиной заболевания является мутация в гене ALPL, кодирующем указанный фермент. Этот ген расположен на коротком плече первой хромосомы, которая содержит большее количество генетической информации о структуре организма человека по сравнению с остальными хромосомами.
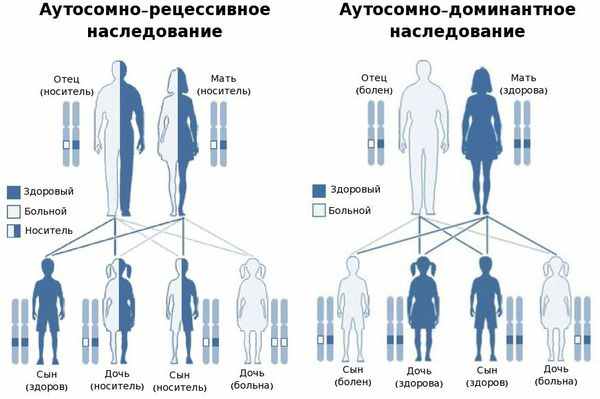
В большинстве случаев заболевание передаётся по аутосомно-рецессивному типу, т. е. при условии, что мутировавший ген передался к ребёнку от обоих родителей. Также описаны и случаи аутосомно-доминантного типа наследования — передача мутировавшего гена только от одного родителя. [25]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы гипофосфатазии
Клиническая картина заболевания в значительной степени зависит от возраста пациента и формы заболевания. [1]
К основным проявлениям гипофосфатазии относят повреждения скелета в виде:
- значительной гипоминерализации костей — нарушения развития костной ткани;
- краниосиностоза — деформации черепа в связи с преждевременным закрытием черепных швов;
- рахитоподобной деформации скелета;
- остеомаляции — ухудшение прочности костей в связи с их недостаточной минирализацией;
- незаживающих переломов;
- повторяющихся переломов, требующих применения инвалидного кресла и других способов поддержки для передвижения. [23]
Боли в суставах, мышцах и костях — одно из значительных и прогрессирующих проявлений гипофосфатазии. [14]
Поражение дыхательной системы проявляется гипоплазией (недоразвитием) лёгких и дыхательной недостаточностью, при которой в некоторых случаях требуется применение кислородной поддержки. [23]
Со стороны центральной нервной системы наблюдаются судорожные приступы, повышение внутричерепного давления, различные внутричерепные кровотечения. [20]
Очень часто при гипофосфатазии встречаются тяжёлые поражения почек в виде нефрокальциноза и почечной недостаточности.
Патология мышечной системы проявляется различными миопатиями, хронической болью в мышцах, задержкой или полным отсутствием моторного развития. [22]
Характерны поражения суставов в форме хондрокальциноза (псевдоподагры) и прогрессирующего артрита.
Гипоминерализация костей приводит к раннему выпадению зубов у детей и потере здоровых зубов у взрослых. [20]
Ещё одним характерным проявлением гипофосфатазии является отставание костного возраста от паспортного по результатам рентгенологического исследования.
При перинатальной форме гипофосфатазии отмечается укорочение трубчатых костей, недоразвитие и укорочение рёбер, мягкие кости черепа, повышается вероятность поражения центральной нервной системы в процессе родов. [20]
Патогенез гипофосфатазии
Патогенез гипофосфатазии связан со снижением выработки либо активности щелочной фосфатазы, структура и функционирование которой нарушается п ри наличии мутации в гене ALPL .
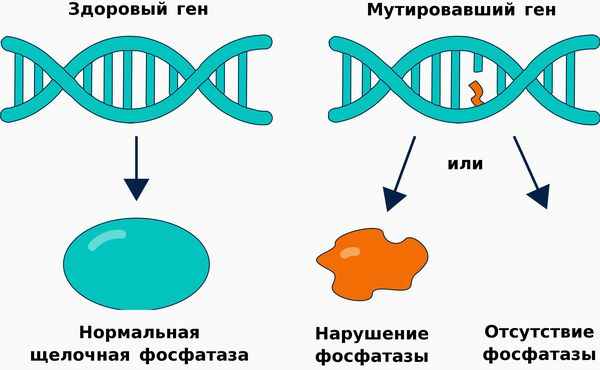
Щелочная фосфатаза принимает участие в метаболизме трёх важных субстратов:
- пиридоксаль-5-фосфат (витамин В6 , PLP);
- неорганический пирофосфат (PPi);
- фосфоэтаноламин (PEA).
Нарушение метаболизма пиридоксаль-5-фосфата приводит к повышению его циркуляции в крови и недостатку активной формы витамина В6. В циркулирующей форме витамин В6 не может проникнуть через гемато-энцефалический барьер. Недостаток активной формы витамина сопровождается поражением центральной нервной системы, что проявляется развитием судорожного синдрома, снижением мозговой активности и трудностями в усвоении информации. [23]
Неорганический пирофосфат является основным компонентом гидроксиапатита, входящего в состав костей. При нарушении метаболизма неорганического пирофосфата происходит повышение его концентрации в крови, гиперфосфатемия и гиперкальциемия, что приводит к отложению солей кальция в суставах, почках и других внутренних органах:
- отложение солей кальция в суставах является причиной артритов, в том числе, тяжёлых;
- поражение почек приводит к развитию почечной недостаточности и полиорганным нарушениям.
Неорганический пирофосфат накапливается и в швах черепа, что сопровождается развитием краниосиностоза, нарушением роста головного мозга, родовым травматизмом, внутричерепной гипертензией, мозговыми кровоизлияниями и отёком диска зрительного нерва. [23]
В результате этих процессов снижается содержание гидроксиапатита в костях, что приводит к деминерализации костей, их искривлению, укорочению и переломам. Патология цементирования зубов является причиной их раннего выпадения. [16] Из-за деформации грудной клетки развивается дыхательная недостаточность и гипоплазия лёгких. Снижение плотности и прочности костей черепа приводит к травмам головного мозга. Искривления позвоночника вызывают сдавление спинного мозга и нарушение осанки. [22]
Фосфоэтаноламин является промежуточным метаболитом, нарушение его метаболизма сказывается на различных реакциях.
Классификация и стадии развития гипофосфатазии
Выделяют четыре формы гипофосфатазии:
- пренатальная (предродовая);
- инфантильная;
- детская;
- взрослая.
При пренатальной форме клинические проявления развиваются ещё до родов и характеризуется задержкой внутриутробного роста плода, гипоксией, дефектами черепа ("мембранозный череп"), деформациями костей, тяжёлой патологией грудной клетки и гипоплазией лёгких. Такие дети в большинстве случаев погибают внутриутробно либо рождаются преждевременно, у них развивается дистресс-синдром (тяжёлое расстройство дыхания). [20] Ранее считалось, что 100% таких детей погибает, но в последнее время благодаря своевременной диагностике и лечению возможно выхаживание пациентов с пренатальной формой гипофосфатазии и продление их жизни. Данную форму заболевания можно диагностиравать во время ультразвукового исследования в период беременности по характерным эхографическим признакам. Тип наследования: аутосомно-рецессивный. [25]
Инфантильная форма проявляется в скором времени после рождения. Смертность таких пациентов без лечения достигает 40%. В неонатальном периоде дети с этой формой гипофосфатазии возбудимы, им характерен плохой аппетит, наблюдается повышенная реакция на внешние раздражители, судороги, диспепсия (нарушение пищеварения), обезвоживание, снижение мышечного тонуса, тяжёлые костные аномалии (мягкие кости, увеличенные черепные швы и роднички), повышение кальция в крови. Часто смерть наступает в неонатальном периоде в связи с тяжёлой дыхательной недостаточностью, но известны случаи спонтанного улучшения. [17] Деформация грудной клетки в 50% случаев приводит к развитию пневмонии . В 64% случаев пациентам требуется кислородная поддержка, 66% детей с этим заболеванием нуждаются в искусственной вентиляции лёгких, из которых 95%, к сожалению, умирают. [17] Тип наследования: аутосомно-рецессивный. [25]
При детской форме гипофосфатазии наблюдается постепенное развитие рахитоподобных скелетных изменений, ранняя потеря зубов, хондрокальциноз и артропатии. При данной форме заболевания очень часто ошибочно ставится диагноз "Рахит", что приводит к неправильному лечению и развитию осложнений. Тип наследования: аутосомно-рецессивный. [25]
При взрослой форме наблюдаются частые переломы и снижение минеральной плотности костей, [7] выпадение зубов, искривление конечностей, встречается изолированная фосфоэтаноламинурия. При рентгенографии выявляется уменьшение минеральной плотности костей черепа, трубчатых костей и отложение кальция в почках. Тип наследования: аутосомно-доминантный. [25]
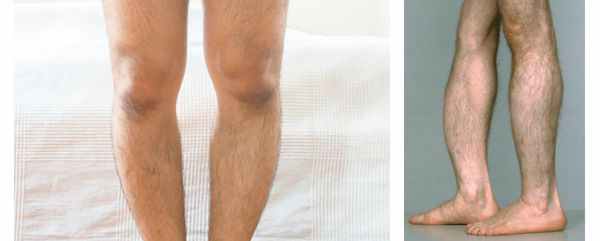
Некоторые авторы выделяют отдельно зубную форму гипофосфатазии . Она характеризуется выпадением здоровых зубов и отсутствием другой симптоматики. Считается самым лёгким вариантом течения гипофосфатазии. [22]
Осложнения гипофосфатазии
Гипофосфатазия является жизнеугрожающим состоянием.
В раннем детском возрасте основными осложнениями заболевания, приводящими к смерти, являются дыхательная недостаточность, травмы головного мозга, внутримозговые кровоизлияния. [12]
В более старшем возрасте развивается почечная недостаточность, связанная с нефрокальцинозом (отложением солей кальция в почках). Это приводит к полиорганной недостаточности и смерти.
Инвалидизация пациентов происходит за счёт снижения мышечного тонуса и артропатий. Поэтому многие пациенты нуждаются в специальных устройствах для облегчения передвижения, в том числе, в инвалидных креслах. [22]

Недостаток биодоступного витамина В6 вызывает судороги, снижение когнитивных функций, задержку психического развития. [8] Эти осложнения лечат витаминами группы В, однако это приводит к избытку пиридоксаль-5-фосфата в крови.
Краниосиностоз приводит к нарушению роста головного мозга, повышению внутричерепного давления, многим пациентам требуется оперативное лечение.
Очень часто осложнения наступают по причине неправильно поставленного диагноза и соответствующего лечения. Так, детям с гипофосфатазией во многих случаях ошибочно ставится диагноз "Рахит" и проводится лечение витамином Д и препаратами кальция. Это приводит к гиперкальциемии, усиленному отложению солей в суставах и почках, а также к гипервитаминозу Д.
Диагностика гипофосфатазии
Диагностика гипофосфатазии основывается на выявлении снижения щелочной фосфатазы в крови.
Диагностика пренатальной и инфантильной гипофосфатазии
Диагностировать пренатальную форму гипофосфатазии возможно во время беременности. Ей характерны следующие ультразвуковые признаки:
- отсутствие оссификации костей (их окостенения);
- более чёткая визуализация структур головного мозга;
- положительный "тест надавливания"; [5]
- недостаточная оссификация, укорочение и деформация длинных трубчатых костей;
- специфическая картина метафиза (участка трубчатой кости);
- характерные акустические дорожки на ультразвуковом снимке. [22][24]
Отсутствие оссификации костей черепа и "мембранозный череп" являются проявлениями, общими для несовершенного остеогенеза и гипофосфатазии, поэтому необходима дифференциальная диагностика этих состояний. [17]
При ультразвуковом исследовании , как правило, трудно дать количественную оценку остеогенеза. Однако если на картине УЗИ внутренняя структура черепной коробки и поверхность мозга, которые обычно невидны у нормально развивающегося плода, визуализируются ярче обычного, то это можно принять в качестве особенности нарушенной формирования костей черепа. [24]
Далее, если провести "тест надавливания" — сильно нажать датчиком ультразвука на брюшную стенку матери — и посмотреть, насколько деформируется череп плода, то у нормально развивающегося малыша форма черепа сохранится (так как кости черепа твёрдые), а в случае выявленного нарушенного остеогенеза костей черепа и "мембранозного черепа" будет наблюдаться его деформация. [24]
Обычно считается, что для того, чтобы увидеть состояние костей, лучше прибегнуть не к ультразвуковой, а к рентгенологической диагностике . На рентгенограмме ясно видны укорочение и искривление длинных трубчатых костей, а также просветление "языков", которые выступают из метафиза в сторону диафиза, являющееся наиболее специфической диагностической картиной при гипофосфатазии. [24]
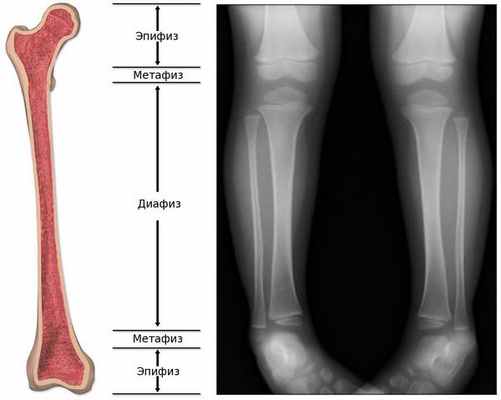
С другой стороны, на ультразвуковом снимке можно убедиться в таких особенностях, как укорочение длинных трубчатых костей, загибы и деформация, но при этом трудно определить, является ли искривление следствием перелома кости или изгиба, а также трудно визуализировать просветление пониженной плотности ("языки пламени" у более взрослых пациентов). [24]
При гипофосфатазии происходит относительно хорошее окостенение средней части диафиза (тела) длинных трубчатых костей, однако к периферии костей оссификация постепенно ухудшается, и на участках метафиза видна только поверхность костей. Поэтому на рентгенограмме это визуализируется как просветление "языков" пониженной плотности. [13]
На ультразвуковом снимке относительно окостеневшие средняя и периферическая части диафиза визуализируются как линии низкой плотности. [10] С другой стороны, рядом с метафизом на поверхности наблюдается незначительная оссификация, изображение поверхности кости нечёткое, под диафизом с обеих сторон появляются "пояски". Всё это является специфической ультразвуковой картиной недостаточно "окрепших" костей при гипофосфатазии и не наблюдается при нормальном развитии плода. [24]
Описание характерных акустических дорожек
Ультразвуковой луч, прошедший через мягкие ткани и достигший поверхности кости, на 90% отражается от её поверхности, что отображается на ультразвуковом снимке в качестве линии низкой эхогенности (плотности). Однако в перинатальном периоде эпифиз практически не заметен и даже у нормально развивающегося плода не визуализируется на ультразвуковом снимке.
В основном у льтразвуковой луч отражается от поверхности кости, а сквозь кость проходит лишь его незначительное количество, поэтому обычно непосредственно под костью визуализируется большая область чёрного цвета, кроме которой больше ничего не видно. Это явление называется "акустическая тень". [18]
На современном оборудовании для ультразвуковых исследований, чтобы визуализировать на снимке область чёрного цвета, находящуюся непосредственно под костью, определяют сильную отражающую волну, после чего автоматически усиливают возвращающийся ультразвуковой сигнал и пытаются визуализировать структуру, которая скрывается в акустической тени. [2] На ультразвуковом снимке длинных трубчатых костей при гипофосфатазии с обеих сторон диафиза в нижней части видны дорожки. Предполагается, что это происходит из-за того, что усиливается ультразвуковой сигнал от метафиза, через который проходит относительно большое количество ультразвуковых лучей. [21]
Диагностика детской и взрослой гипофосфатазии
В более старшем возрасте диагноз можно заподозрить по клиническим проявлениям.
При подозрении на гипофосфатазию определяется уровень щелочной фосфатазы в крови. Приняты следующие нормы щелочной фосфатазы:
Рентгенограмма костей при гипофосфатазии
Рентгенограмма костей при гипофосфатазии
а) Визуализация:
1. Общая характеристика:
• Лучший диагностический критерий:
о Перинатальная: тяжелая гипоминерализация скелета
о Инфантильная: тяжелые рахитоподобные изменения
о Детская: Рахитоподобные изменения различной тяжести
о Взрослая: изменения, сходные с остеомаляцией
2. Рентгенография при гипофосфатазии:
• Перинатальная летальная форма:
о Значительно сниженная минеральная плотность кости
- Скелет может выглядеть полностью неминерализованным
о Микромелия
о Остемаляция, приводящая к деформациям:
- Деформации грудной клетки → затруднения дыхания → смерть
о Костные шпоры Боулдера в диафизах локтевых и малоберцовых костей
о Ложное расхождение швов черепа с функциональным краниосиностозом
• Перинатальная доброкачественная форма:
о Скелетные изменения такие же, как при летальном типе
о Самостоятельное восстановление скелетных нарушений
• Инфантильная форма:
о Тяжелые рахитоподобные изменения
о Рентгенонегативные отростки распространяются в метафизы из пластинок роста
о Ложное расхождение швов черепа с функциональным краниосиностозом
о Изменение формы тел позвонков
о Низкорослость
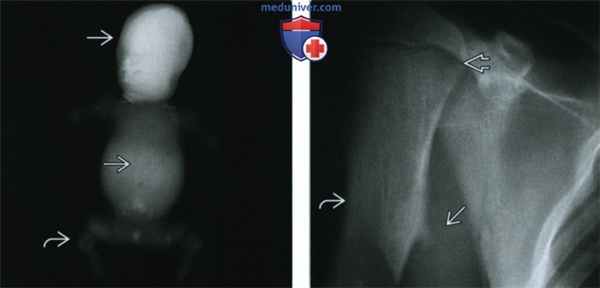
(Слева) Рентгенография в ПЗ проекции, мертворожденный младенец с крайне тяжелой летальной формой гипофосфатазии. Обратите внимание на практически полное отсутствие минерализации скелета и выраженную микромелию.
(Справа) Рентгенография плечевой кости в ПЗ проекции с типичными для детской формы гипофосфатазии изменениями. Отмечается небольшая угловая деформация диафиза плечевой кости и экзостоз. Подобные выросты, характерные для большеберцовой и локтевой костей, могут развиваться в любой кости. Отмечается незначительное расширение пластинки роста.
• Детская форма:
о Низкорослость
о Рахитоподобные изменения
о Шпоры Боулдера
о Ложное расхождение швов черепа с функциональным краниосиностозом
• Взрослая форма:
о Остеопения, изменения, сходные с остеомаляцией
о Псевдопереломы в латеральном проксимальном отделе компактного вещества бедренной кости:
- Псевдопереломы на фоне остеомаляции в компактном веществе медиальной поверхности
о ± отложения пирофосфата кальция
о Множественные стрессовые переломы, особенно в стопах
о Шпоры Боулдера, особенно в локтевой и малоберцовой костях
о Функциональный краниосиностоз
• Одонтогипофосфатазия: изолированное поражение зубов:
о Ранняя потеря молочных зубов
о Аномальное развитие коренных зубов
о Подвижность коренных зубов
3. УЗИ при гипофосфатазии:
• Пренатальное ультразвуковое исследование: III триместр:
о Четкая детализация головного мозга: ↓ минерализации черепа ↑ проведение ультразвука
о Микромелия
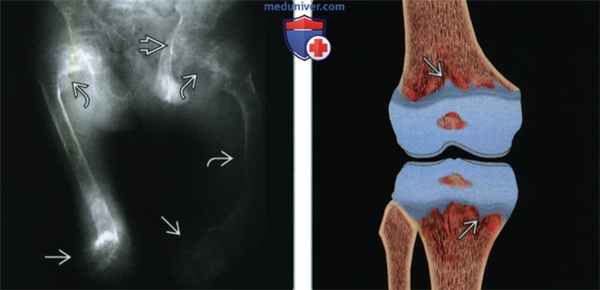
(Слева) Рентгенография в ПЗ проекции: визуализируются обе бедренные кости у ребенка с тяжелой формой гипофосфатазии. Имеются выраженные изменения пластинок роста (расширение и неровность контуров). Остеомаляция осложнилась угловыми деформациями в проксимальных отделах обеих бедренных костей, а также в диафизе левой бедренной кости. Имеются протрузионные деформации тазобедренных суставов, характерные для остеомаляции.
(Справа) На рисунке во фронтальной плоскости показана гипофосфатазия, характеризующаяся выраженной неровностью контуров пластинок роста и отростками суставных хрящей, распространяющимися в метафизы.
б) Дифференциальная диагностика гипофосфатазии:
1. Несовершенный остеогенез:
• Множественные переломы приводят к асимметричному укорочению конечностей; слабая оссификация черепа
2. Остеомаляция и рахит:
• Не столь выраженные деформации и гипоминерализация.
3. Ахондрогенез:
• Отсутствие минерализации преимущественно в позвонках, практически нормальная минерализация в добавочном скелете и костях черепа
в) Патология:
1. Общая характеристика:
• Этиология:
о Мутация гена ALPL → дефицит синтеза ТНЩФ (TNSALP, тканевая неспецифическая щелочная фосфатаза):
- Недостаточный гидролиз неорганического фосфата, накопление которого препятствует образованию гидроксиапатита (минерализованной кости)
• Генетические факторы:
о Инфантильная и перинатальная форма: аутосомно-рецессивные
о Менее тяжелые формы: аутосомно-рецессивные или доминантные
• Сопутствующие нарушения:
о Инфантильная форма: гиперкальциемия, обусловленная неспособностью включения кальция в кость
2. Микроскопия:
• Увеличение остеоидной ткани и неминерализованного хряща
• Пластинка роста: разрушение гипертрофической зоны, наблюдаемое при рахите, кальцинированная зона может полностью отсутствовать
г) Клинические особенности:
1. Проявления:
• Типичные признаки/симптомы:
о Проявления зависят от формы
о При всех формах отмечается ранняя потеря молочных зубов о Перинатальная летальная: мертворождение или смерть в первые несколько дней/недель вследствие нарушений дыхания
о Инфантильная: манифестирует в первые 6 месяцев жизни, симптомы обусловлены гиперкальциемией:
- Раздражительность, алиментарная недостаточность, рвота, нефролитиаз и почечная недостаточность, судороги
о Детская форма: задержка начала ходьбы, ковыляющая походка, боли в костях, проблемы с дыханием вследствие деформаций ребер
о Взрослая форма: боли в костях, деформации скелета, переломы
2. Демография:
• Пол: тяжелые формы: М = Ж
• Эпидемиология: тяжелые формы: 1:100000 новорожденных
3. Течение и прогноз:
• Перинатальная летальная: мертворождение или смерть в первые дни/недели
• Инфантильная: ограниченная продолжительность жизни
• Детская: возможно спонтанное разрешение и рецидив во взрослом возрасте
• Детская и взрослая: деформации и переломы различной выраженности
4. Лечение:
• Только симптоматическое: невозможность адекватного воздействия на основные пути метаболизма
о Проходит испытания заместительная ферментная терапия
д) Список использованной литературы:
1. Bianchi ML: Hypophosphatasia: an overview of the disease and its treatment. Osteoporos Int. 26(12):2743-57, 2015
Неврологические и нейрохирургические аспекты гипофосфатазии
Гипофосфатазия — редкое наследственное прогрессирующее заболевание, вызванное мутацией в гене ALPL, вследствие которой угнетается активность щелочной фосфатазы. Из-за нарушения процесса минерализации костной ткани в клинической картине преобладают рахитоподобные деформации скелета, но зачастую возникают и другие системные проявления — нарушение дыхания, поражение мочевыделительной системы и неврологические расстройства. У пациентов выявляют судороги, задержку физического и психомоторного развития, дефицит внимания, мышечную слабость, быструю утомляемость, внутричерепную гипертензию, связанную с развитием краниосиностозов. Тяжесть гипофосфатазии зависит от времени ее манифестации: наибольшая смертность регистрируется при перинатальной и инфантильной формах заболевания. Диагностика основана на выявлении характерных клинических симптомов — задержки роста и развития, деформации скелета, болей в мышцах и суставах, преждевременного выпадения зубов. В лабораторных анализах отслеживается стойкое снижение уровня щелочной фосфатазы с учетом возраста и пола пациента; при низкой активности фермента уровни субстратов щелочной фосфатазы пиридоксаль-5-фосфат в крови и фосфоэтаноламина в моче всегда повышены. На рентгенограммах длинных трубчатых костей обнаруживаются «языки» просветления, проецирующиеся от зоны роста в метафизы, а также гипоминерализация, остеопения и другие деформации. Все пациенты с подозрением на гипофосфатазию должны быть проконсультированы клиническим генетиком и обследованы на выявление мутации в гене ALPL.
Авторы декларируют отсутствие конфликтов интересов, связанных с публикацией настоящей статьи.
Ключевые слова
Об авторах
Кузенкова Людмила Михайловна, доктор медицинских наук, профессор, заведующая отделением, отделение психоневрологии и психосоматической патологии НИИ педиатрии
Список литературы
1. Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x.
3. Coburn SP, Whyte MP. Role of phosphatases in the regulation of vitamin B-6 metabolism in hypophosphatasia and other disorders. In: Leklem JE, Reynolds RD, editors. Clinical and physiological applications of vitamin B-6. New York, USA: AR Liss; 1988. pp. 65–93.
4. Surtees R, Mills P, Clayton P. Inborn errors affecting vitamin B6 metabolism. Future Neurol. 2006;1(5):615–620. doi: 10.2217/14796708.1.5.615.
5. Baumgartner-Sigl S, Haberlandt E, Mumm S, et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007;40(6):1655–1661. doi: 10.1016/j.bone.2007.01.020.
6. Belachew D, Kazmerski T, Libman I, et al. Infantile hypophosphatasia secondary to a novel compound heterozygous mutation presenting with pyridoxine-responsive seizures. JIMD Rep. 2013;11:17–24. doi: 10.1007/8904_2013_217.
7. Yamamoto H, Sasamoto Y, Miyamoto Y, et al. A successful treatment with pyridoxal phosphate for West syndrome in hypophosphatasia. Pediatr Neurol. 2004;30(3):216–218. doi: 10.1016/j.pediatrneurol.2003.08.003.
8. Fukazawa M, Tezuka J, Sasazuki M, et al. Infantile hypophosphatasia combined with vitamin B6-responsive seizures and reticular formation lesions on magnetic resonance imaging: a case report. Brain Dev. 2018;40(2):140–144. doi: 10.1016/j.braindev.2017.07.015.
9. van Karnebeek CD, Jaggumantri S. Current treatment and management of pyridoxine-dependent epilepsy. Curr Treat Options Neurol. 2015;17(2):335. doi: 10.1007/s11940-014-0335-0.
10. Diez-Zaera M, Diaz-Hernandez JI, Hernandez-Alvarez E, et al. Tissue-nonspecific alkaline phosphatase promotes axonal growth of hippocampal neurons. Mol Biol Cell. 2011;22(7):1014–1024. doi: 10.1091/mbc.E10-09-0740.
11. Mornet E. Hypophosphatasia. Metabolism. 2018;82:142–155. doi: 10.1016/j.metabol.2017.08.013.
15. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233– 246. doi: 10.1038/nrendo.2016.14.
16. Durussel J, Liu J, Campbell C, et al. Bone mineralizationdependent craniosynostosis and craniofacial shape abnormalities in the mouse model of infantile hypophosphatasia. Dev Dyn. 2016;245(2):175–182. doi: 10.1002/dvdy.24370.
17. Poryo M, Meyer S, Eymann R, et al. Clinical images: a cloudy skull — hypophosphatasia as reason for copper-beaten skull. Neuropediatrics. 2016;47(6):410–411. doi: 10.1055/s-0036- 1593532.
18. Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep. 2016;14(3):95–105. doi: 10.1007/s11914-016-0309-0.
19. Баранов А.А., Намазова-Баранова Л.С., Савостьянов К.В., и др. Клинические рекомендации по диагностике и лечению гипофосфатазии у детей // Педиатрическая фармакология. — 2016. — Т.13. — №6 — С. 539–543. [Baranov AA, NamazovaBaranova LS, Savostianov КV, et al. Clinical recommendation to the diagnostics and treatment of hypophosphatasia in children. Pediatric pharmacology. 2016;13(6):539–543. (In Russ).] doi: 10.15690/pf.v13i6.1665.
20. Kishnani PS, Rush ET, Arundel P, et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol Genet Metab. 2017;122(1–2):4–17. doi: 10.1016/j. ymgme.2017.07.010.
23. Mornet E, Yvard A, Taillandier A, et al. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75(3):439–445. doi: 10.1111/j.1469-1809.2011.00642.x.
Гипофосфатазия: как заподозрить заболевание у ребенка? Клинические наблюдения
Цель статьи: описать особенности дифференциальной диагностики гипофосфатазии с рахитоподобными заболеваниями на примере нескольких клинических случаев.
Основные положения. Причиной гипофосфатазии является снижение активности щелочной фосфатазы (ЩФ), приводящее к поражению всех органов и тканей различной степени тяжести с многообразными проявлениями. Неспецифическая клиническая картина редкого наследственного метаболического заболевания, низкая осведомленность врачей о проблеме определяют трудности диагностики гипофосфатазии. Единственный метод патогенетического лечения гипофосфатазии — пожизненная ферментная заместительная терапия препаратом асфотаза альфа. В статье приведены клинические случаи перинатальной (более тяжелой) и детской формы заболевания.
Заключение. Наличие гипофосфатазии следует проверять у всех пациентов с признаками гипоминерализации костей в сочетании с полиорганными нарушениями и/или судорожным синдромом, патологией зубочелюстного аппарата и снижением активности ЩФ. Важно отметить, что показатели активности ЩФ имеют возрастные особенности, следовательно, лечащий врач должен убедиться, что сообщаемые лабораторией результаты отражают нормальный для пациента конкретного возраста уровень ЩФ.
Вклад авторов: Храмова Е.Б. — разработка дизайна статьи, проверка критически важного содержания, написание текста рукописи, утверждение рукописи для публикации; Левитина Е.В., Романенко Е.С. — обследование и лечение пациентов, написание текста рукописи; Супрунец С.Н., Гуркина Е.Ю. — обследование и лечение пациентов; Кучкина А.Ю. — обзор публикаций по теме статьи, написание текста рукописи.
Конфликт интересов: авторы заявляют об отсутствии возможного конфликта интересов.
В отечественных и зарубежных научных публикациях подробно описаны классификация, разнообразие клинических симптомов, принципы лабораторной диагностики и результаты молекулярно-генетических исследований при гипофосфатазии [1–4] . Очевидно, что гипофосфатазия является прогрессирующим наследственным метаболическим заболеванием, вызванным дефицитом щелочной фосфатазы (ЩФ), который возникает из-за мутации в гене ALPL , картированном на 1 хромосоме (1p36.12), кодирующем изофермент тканенеспецифичной ЩФ (ТНЩФ).
Патогенез гипофосфатазии достаточно хорошо изучен. В норме фермент ТНЩФ непосредственно влияет на отщепление фосфатной группы от неорганического пирофосфата, высвобождающийся неорганический фосфат связывается с кальцием, формируя кристаллы гидроксиапатита, необходимые для минерализации костного матрикса. В результате дефицита активности ТНЩФ неорганический пирофосфат не расщепляется и кристаллы гидроксиапатита не образуются, что, безусловно, приводит к нарушению минерализации костной ткани. В свою очередь, накапливающийся в плазме и тканях неорганический пирофосфат соединяется с аморфным фосфатом кальция с образованием кристаллов пирофосфата кальция, это вызывает нефрокальциноз или становится причиной артрита [5] .
Еще одной крайне важной функцией ТНЩФ является отщепление фосфора от пиридоксаль-5-фосфата, что делает возможным проникновение пиридоксаля через клеточные мембраны в ЦНС, где происходит повторное присоединение фосфата к пиридоксалю. Вновь образованный пиридоксаль-5-фосфат выступает в роли кофактора многих нейротрансмиттеров, а его дефицит в ЦНС приводит к развитию пиридоксин-зависимых судорог [6] .
В зависимости от возраста дебюта гипофосфатазии выделяют перинатальную форму (появление симптомов уже внутриутробно или сразу после рождения); младенческую, или инфантильную (появление симптомов в первые 6 месяцев жизни); детскую и взрослую формы (появление клинических симптомов соответственно до 18 лет и позже). Форма заболевания во многом определяет и тяжесть его течения — от 100% летальной (в отсутствие терапии) перинатальной формы до относительно легкого течения при взрослой форме.
Тяжелые формы гипофосфатазии развиваются, как правило, при наличии гомозиготной или компаунд-гетерозиготной мутации в гене ALPL . ЩФ экспрессируется на поверхности клеток как гомодимер, поэтому некоторые гетерозиготные мутации могут снижать активность всего гомодимера, приводя к доминантно негативному эффекту. Вследствие этого наличие мутации даже в одной аллели может провоцировать развитие заболевания.
У носителей одинаковой мутации в семье возможна различная степень тяжести заболевания, что указывает на наличие модулирующих факторов.
В некоторых случаях гипофосфатазии не удается обнаружить мутации в гене ALPL , поэтому для верификации диагноза ведущими критериями являются клинические признаки заболевания и снижение активности ЩФ ниже нормы для данного возраста и пола [7] .
Единственный метод патогенетического лечения гипофосфатазии — пожизненная ферментная заместительная терапия препаратом асфотаза альфа, которая представляет собой человеческий рекомбинантный тканенеспецифический химерный Fc-дека-аспартатный гликопротеин ЩФ. По данным литературы, применение асфотазы альфа при перинатальной форме гипофосфатазии способствует лучшей выживаемости пациентов по сравнению с таковой в группе контроля: 95% против 42% в возрасте 1 года, 84% против 27% в возрасте 5 лет соответственно (р < 0,0001 в модели Каплана — Мейера, оценивающей долю пациентов, проживших какое-либо время после приема некого лекарственного препарата) [7] .
Приводим описание клинических случаев гипофосфатазии у детей.
КЛИНИЧЕСКИЙ СЛУЧАЙ 1
Мальчик Ч. переведен из ГБУЗ ТО «Перинатальный центр» (г. Тюмень) в отделение патологии новорожденных ГБУЗ ТО ОКБ № 2 (г. Тюмень) в возрасте 5 дней с диагнозом: Транзиторное тахипноэ у новорожденного. Врожденный порок развития — дефект костей черепа, аплазия теменных и височных костей. Гипофосфатазия? — для верификации диагноза и определения тактики терапии.
Из анамнеза жизни и заболевания известно, что ребенок родился от первых самостоятельных родов в сроке 38,3 недели, в головном предлежании, с оценкой по шкале Апгар 7–7 баллов. При рождении масса тела — 3570 г, длина — 54 см, окружность головы — 35 см, окружность грудной клетки — 34 см.
При УЗИ плода в сроке 29,6 недели диагностирована гипоплазия костей носа. При рождении обращали на себя внимание скелетные диспропорции, укорочение и деформация конечностей; грудная клетка уплощена, отсутствуют кости мозгового черепа (мембранозный череп). Через 2 часа после рождения отмечено тахипноэ, потребовавшее кратковременной респираторной поддержки.
На момент поступления в отделение патологии новорожденных состояние ребенка среднетяжелое, сознание ясное. Конечности укорочены, деформированы за счет внутриутробных переломов трубчатых костей, мембранозный череп, грудная клетка уплощена, обе половины грудной клетки симметрично участвуют в акте дыхания. Респираторных нарушений нет, частота дыхательных движений — 42 в мин. Сатурация кислородом — 98%. Диффузная мышечная гипотония, гипорефлексия, снижена двигательная активность. ЧСС — 148 в мин. Вскармливание грудное, объем питания по возрасту, усваивает.
Наиболее значимые для диагностики заболевания результаты обследования
Уровень фосфора — 2,21 ммоль/л (норма — 1,45–2,16 ммоль/л), кальция общего — 2,36 ммоль/л (норма — 2,2–2,5 ммоль/л), ЩФ — 28 Ед/л (норма — 53–128 Ед/л). Отмечаются стойкое повышение содержания фосфора и снижение уровня ЩФ в динамике, нарастание концентрации кальция в сыворотке до 3,1 ммоль/л, ионизированного кальция — до 1,7 ммоль/л. Содержание паратгормона, витамина D — в пределах референсных интервалов.
Рентгенография костей скелета: лобная кость представлена двумя пластинами, две пластины височных костей, множественные переломы ребер с обеих сторон, перелом грудинной трети правой ключицы без смещения, переломы дистальной и проксимальной третей плечевой, лучевой и локтевой кости справа и слева, переломы проксимального и дистального метаэпифиза обеих костей правой и левой голени. Деформированные седалищные кости, крылья и тело подвздошных костей. Трубчатые кости деформированы, со слабой периостальной костной реакцией, с грубой кистовидной перестройкой структуры. Ростковые зоны костей не определяются ( рис. 1 ).
Рис. 1. Рентгенография костей скелета и черепа пациента Ч. Фото авторов
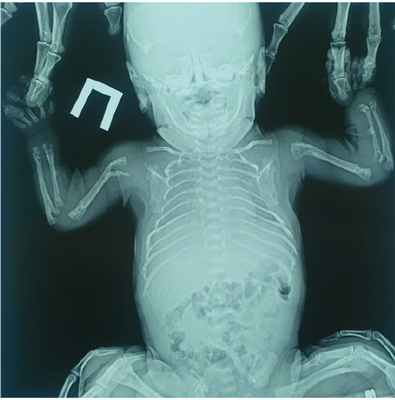
Пациенту выполнено молекулярно-генетическое исследование (метод секвенирования нового поколения и секвенирование по Сэнгеру), выявлен вариант нуклеотидной последовательности c.1171delC в 10 экзоне гена ALPL (chr1:g.21902393AC>A; rs779683021) в гетерозиготном состоянии, также вариант нуклеотидной последовательности в 5 экзоне c.314C>T (chr1:g.21889619C>A; rs768348242) в гетерозиготном состоянии.
На основании клинико-лабораторного и молекулярно-генетического обследования верифицирован диагноз: Гипофосфатазия, перинатальная форма. Аплазия теменных костей, гипоплазия лобных, височных и затылочных костей. Множественные патологические переломы трубчатых костей, ребер, ключиц.
При обследовании матери пациента определен низкий уровень ЩФ — 32 Ед/л, аналогично и у отца — 20 Ед/л (норма — 40–150 Ед/л). Родители не имеют клинических проявлений метаболического заболевания, брак не родственный.
При молекулярно-генетическом обследовании матери пробанда выявлен патогенный вариант c.1171delC (p.Arg391ValfsTer12) в гетерозиготном состоянии в гене ALPL; у отца пробанда найден, вероятно, патогенный вариант c.302A>G (p.Ala105Val) в гетерозиготном состоянии в гене ALPL .
Обследование ребенка и родителей выполнялось в генетической лаборатории сектора клинико-генетических исследований Организационно-методического отдела по медицинской реабилитации ГБУЗ «Городская больница № 40» (начальник сектора — к. б. н. Глотов О.С.), г. Санкт-Петербург.
Пациенту назначена фермент-заместительная терапия препаратом асфотаза альфа в дозе 2 мг/кг подкожно 3 раза в неделю, переносит терапию удовлетворительно, нежелательные явления не отмечаются. Продолжено динамическое наблюдение.
КЛИНИЧЕСКИЙ СЛУЧАЙ 2
Девочка У. с 5 месяцев наблюдалась у невролога с мышечной слабостью, проходила курсы массажа и физиотерапии с незначительным положительным эффектом. Из анамнеза жизни: голову удерживает с 3 месяцев, сидит с 10 месяцев, ползает с 12 месяцев, ходит с поддержкой с 1 года 3 месяцев, к 18 месяцам ходит самостоятельно неуверенно, балансируя.
В связи с отставанием в формировании статико-моторных функций на фоне умеренной мышечной гипотонии с целью исключения рахитоподобного заболевания в возрасте 1 года 2 месяцев впервые определено содержвание ЩФ: оно оказалось сниженным до 98 Ед/л (при норме 156–369 Ед/л). Повторно обследована в 1 год 5 месяцев: уровень ЩФ — 102 Ед/л (норма — 108–317 Ед/л), кальция общего — 2,7 ммоль/л (норма — 1,9–2,6 ммоль/л), кальция ионизированного — 1,33 ммоль/л (норма — 1,12–1,32 ммоль/л), фосфора — 1,9 ммоль/л (норма — 1,29–2,26 ммоль/л), 25(ОН)D — 50 нг/мл (норма — 30–70 нг/мл), пиридоксина — 18,8 мкг/л (норма — 2,2–27,9 мкг/л), остеокальцина — 46,4 нг/мл (норма — 8,4–33,9 нг/мл).
Рентгенография трубчатых костей, кистей рук с захватом лучезапястных суставов: отставание костного возраста на один эпикризный срок с нарушением порядка окостенения, определяются псевдоэпифизы пястных костей, умеренный остеопороз костей голеней. УЗИ органов мочевой системы: утолщение паренхимы почек, большое количество мелкодисперсной взвеси в просвете мочевого пузыря.
Ребенку выполнено молекулярно-генетическое исследование — выявлен вариант нуклеотидной последовательности g.21902399del в гене ALPL в гетерозиготном состоянии (исследование производилось в лаборатории молекулярной генетики и клеточной биологии ФГБУ «НМИЦ здоровья детей» Минздрава России, заведующий лабораторией — к. б. н. Савостьянов К.В.). При обследовании родителей аналогичная мутация найдена у матери ребенка. Таким образом, у пациентки У. диагностирована гипофосфатазия, детская форма.
При осмотре в возрасте 2 лет 5 месяцев сохранялись умеренное снижение мышечного тонуса, неуверенная ходьба, незначительная деформация черепа. Ребенок не бегает, быстро устает, может подниматься по лестнице с поддержкой. Улучшилась мелкая моторика — берет в руку карандаш, стала работать с мелкими предметами.
Речь практически отсутствует, общается с помощью мимики, жестов, ведет за руку. Игровая деятельность не развита, активной сюжетной игры нет, иногда эпизоды вокальных реакций с эмоциональной окраской. Общение не активное, обычно играет одна, реагирует на контакты с близкими людьми, со сверстниками в игру не вступает. Инструкции понимает избирательно, большинство выполняет, чаще действует по подражанию.
Получает симптоматическую терапию. Решение вопроса о назначении фермент-заместительной терапии — при ухудшении состояния в динамике. Прогноз для жизни благоприятный.
КЛИНИЧЕСКИЙ СЛУЧАЙ 3
На момент обращения к детскому эндокринологу пациент Р. (возраст — 6 лет 4 месяца) и пациент А. (возраст — 1 год) имели однотипные жалобы: раннее выпадение молочных зубов без резорбции корня, быстрая утомляемость.
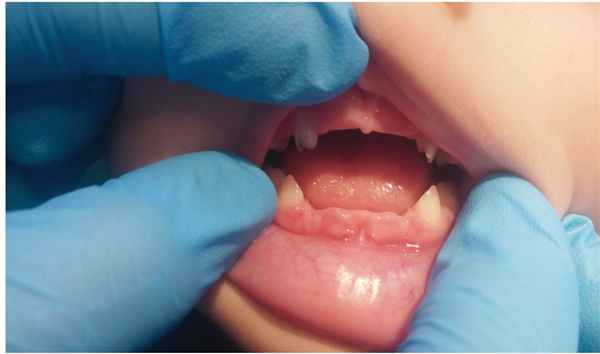
Данные анамнеза жизни и заболевания пациента Р.: ребенок от первой беременности, срочных оперативных родов (ягодичное предлежание плода). Масса при рождении — 3490 г, длина — 51 см, окружность головы — 36 см, оценка по шкале Апгар — 8–8 баллов. С возраста 1 месяца ребенок наблюдался ортопедом по поводу дисплазии тазобедренных суставов. С 12 месяцев у мальчика стали выпадать молочные зубы, появились боли в конечностях и быстрая утомляемость, неустойчивая походка и частые падения. Осмотрен стоматологом в возрасте 13 месяцев, заключение: Генерализованный пародонтит тяжелой степени. В течение второго года жизни продолжалась потеря молочных зубов ( рис. 2 ).
Рис. 2. Выпадение зубов без резорбции корня у пациента Р. в возрасте 2 лет. Фото авторов
К четырем годам пациент неоднократно перенес двусторонний экссудативный средний отит, имел двустороннюю кондуктивную тугоухость 1-й степени, хронический аденоидит. У ребенка сохранялись боли в конечностях, неустойчивая походка и частые падения, сформировалось нарушение осанки.
В 2018 г. в семье родился второй мальчик. Пациент А. от второй беременности, вторых срочных самостоятельных родов, масса тела — 3240 г, длина — 52 см, окружность головы — 36 см, оценка по шкале Апгар — 7–8 баллов. С возраста 1 месяца ребенок наблюдается по поводу врожденного стридора. По данным медицинской документации, в возрасте 3 месяцев у младенца диагностирован рахит легкой степени, назначены курс общего массажа и прием витамина D в дозе 3000 МЕ/сут в течение 1 месяца.
В 11 месяцев ребенок упал с высоты собственного роста, при этом у него выпали 2 нижних резца. Родители обратились к детскому эндокринологу для исключения наследственного заболевания у детей.
Пациентам выполнено комплексное обследование в рамках дифференциальной диагностики нарушений фосфорно-кальциевого обмена, основные результаты представлены в таблице .
Таблица
Результаты обследования пациентов Р. и А., имеющие непосредственное отношение к диагностике заболевания
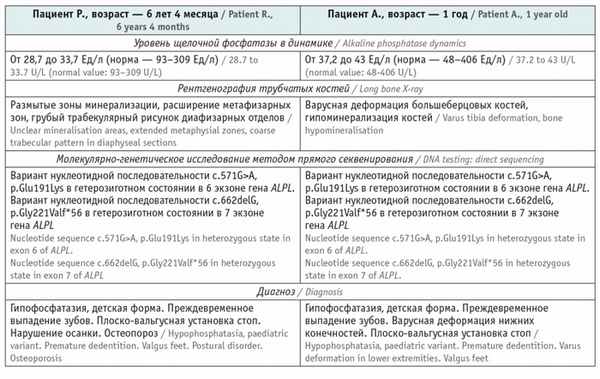
У матери пробандов уровень ЩФ — от 28,4 до 36 Ед/л (норма — 30–120 Ед/л), молекулярно-генетический анализ: методом прямого секвенирования определен вариант нуклеотидной последовательности c.662delG, p.Gly221Valf*56 в гене ALPL в гетерозиготном состоянии. У отца пробандов концентрация ЩФ — 38 Ед/л (норма — 53–128 Ед/л), молекулярно-генетический анализ: методом прямого секвенирования выявлен вариант нуклеотидной последовательности c.571G>A, p.Glu191Lys в гене ALPL в гетерозиготном состоянии. Родители пациентов Р. и А. не имеют клинических проявлений гипофосфатазии. Брак не родственный.
Больным Р. и А. рекомендовано проведение фермент-заместительной терапии препаратом асфотаза альфа в дозе 2 мг/кг массы тела путем подкожной инъекции 3 раза в неделю пожизненно. Продолжено динамическое наблюдение. Прогноз для жизни благоприятный.
Диагностика перинатальной формы гипофосфатазии не представляет особых затруднений. Наличие множественных внутриутробных переломов, деформации скелета, гипоминерализации костей, пиридоксин-зависимых судорог, дыхательной недостаточности и гипоплазии легких в сочетании с низким уровнем ЩФ при нормальных значениях паратгормона, витамина D, нормальном или повышенном уровне кальция в крови позволяют верифицировать диагноз, исключить другие формы хондродисплазий и несовершенный остеогенез.
Определение низкого уровня ЩФ будет отправной точкой в дифференциальной диагностике младенческой и детской форм гипофосфатазии с другими вариантами рахитоподобных заболеваний. Следует помнить, что преждевременное выпадение молочных зубов иногда является первым и даже единственным признаком гипофосфатазии. При этом выпадение зубов происходит без рассасывания корня, сопровождается уменьшением высоты альвеолярной кости и расширением корневых каналов.
Непрогрессирующая проксимальная миопатия может быть ранним признаком гипофосфатазии. Считается, что симптомы могут возникать в результате повышения уровня пирофосфата или из-за других, пока неизвестных факторов, которые способны ингибировать мышечную функцию. У детей с гипофосфатазией может быть «утиная» (ковыляющая) и замедленная походка [5] .
В описанных клинических случаях 2 и 3 признаки гипофосфатазии определялись уже в первом полугодии жизни, в дальнейшем появлялись новые симптомы, однако верификация диагноза произошла только в дошкольном возрасте. Нельзя исключить наличие младенческой формы заболевания в этих случаях.
Следует обратить внимание врачей на обязательную оценку уровня щелочной фосфатазы (ЩФ) как основополагающего лабораторного теста для подтверждения диагноза гипофосфатазии при дифференциальной диагностике метаболических заболеваний костей. Важно отметить, что показатели активности ЩФ имеют возрастные особенности, следовательно, лечащий врач должен убедиться, что сообщаемые лабораторией результаты отражают нормальный для пациента конкретного возраста уровень ЩФ.
Гипофосфатазия
Редкая генетическая болезнь, связанная с нарушением минерального обмена и приводящая к разрушению костей и зубов.
О болезни
Гипофосфатазия — редкое наследственное заболевание, при котором нарушается минерализация костей и зубов. Чем раньше появляются признаки болезни, тем тяжелее она протекает. Возникает в результате мутаций в гене ALPL, который отвечает за минеральный баланс и развитие костной ткани.
Симптомы варьируются от изолированного поражения зубов до тяжелых форм с нарушением развития костей конечностей, черепа, грудной клетки. На данный момент разработано лечение, которое направлено на устранение последствий мутаций.
Подробнее
Механизм возникновения
Гипофосфатазия является результатом мутации в гене ALPL, который отвечает за синтез фермента щелочной фосфатазы костей. Снижение активности фермента ведет к повышению уровня особых веществ — фосфоэтаноламина и неорганического пирофосфата, которые блокируют поступление минералов в костную ткань [1]. Если активность щелочной фосфатазы полностью отсутствует, то развивается тяжелая форма гипофосфатазии, а если снижена — более мягкие варианты.
Гипофосфатазия передается по аутосомно-доминантному или аутосомно-рецессивному типу. При аутосомно-доминантном варианте мутантный ген может передаваться от больного родителя (в 50% случаев) или появляться у пациента спорадически, то есть случайно. При аутосомно-рецессивном типе для развития болезни нужно унаследовать мутацию от каждого родителя.
Симптомы
Чем раньше начались проявления болезни, тем тяжелее её течение. Симптомы гипофосфатазии разнообразны и могут появиться в любом возрасте:
- аномалии скелета, похожие на рахит: размягчение костей, укороченные конечности, деформация грудной клетки и черепа,
- медленный рост и плохой набор веса,
- одышка, тошнота и рвота, запоры, судороги, нарушение функции почек,
- раннее выпадение молочных зубов, аномалии и выпадение коренных зубов, низкий рост, деформация рук и ног, увеличение голеностопных и запястных суставов,
- переломы костей бедра и стопы в результате остеомаляции,
- хронический болевой синдром, снижение двигательной активности, артралгии
Лечение
Симптоматическая терапия для облегчения состояния:
- Для нормализации уровня минералов — диета с низким содержанием кальция и витамина D, достаточное употребление жидкости, диуретики, кальцитонин.
- При тяжелых состояниях — может потребоваться ИВЛ.
- Обезболивающие — препараты из группы НПВС: анальгин, ибупрофен.
- Витамин В6 — для борьбы с судорогами.
- При повышенном внутричерепном давлении — операция шунтирования.
- При искривлении и переломах конечностей — ортопедические конструкции, а при частых переломах трубчатых костей — установка металлических стержней.
Важно отметить, что при гипофосфатазии противопоказано применение препаратов из группы бисфосфонатов, которые применяются при остеопорозе: алендроната, ризедроната, золендроната и некоторых других. Они не только не улучшат состояние костей, но и могут привести к усугублению болезни.
На стадии разработки находятся следующие методы лечения [3]:
- Терипаратид (препарат Фортео). Это паратиреоидный гормон, стимулирующий образование новой костной ткани и показанный пациентам при переломах и трещинах.
- Трансплантация костного мозга. При тяжелом течении гипофосфатазии пациенту пересаживают остеобласты (молодые клетки костной ткани) и стволовые клетки, что способствует образованию здоровой костной ткани.
- Антитела к склеростину (препарат Ромосозумаб). Склеростин — это вещество, которое содержится в клетках костной ткани и регулирует процессы ее образования и разрушения. Антитела к склеростину подавляют его действие, на фоне чего молодые клетки костной ткани (остеобласты) активизируются, и костная масса увеличивается.
Реабилитация
Включает физиопроцедуры, трудотерапию, умеренную физическую активность. Рекомендовано регулярно посещать стоматолога, а при необходимости для передвижения использовать костыли, подпорки, инвалидную коляску.
Прогноз
При легких формах болезни, а также при любых вариантах, которые своевременно диагностировали и начали ферментозаместительное лечение, прогноз благоприятный.
Читайте также: