Селективный IgA дефицит
Добавил пользователь Morpheus Обновлено: 07.01.2026
Для понимания современного состояния проблемы стоматологических проявлений первичных иммунодефицитов проанализирована соответствующая зарубежная литература. В статье описаны стоматологические проявления многих первичных иммунодефицитов, которые могут быть как вторичным, так и основным симптомом. Представлены синдром тяжелого комбинированного иммунодефицита, гипер-IgE синдром, синдромы Вискотта – Олдрича, Ди Джорджи, дефицит STIM1 и дефицит ORAI1, дефицит NEMO и дефицит IκBα, общая вариабельная иммунная недостаточность, X-сцепленная агаммаглобулинемия, гипер-IgM синдром, селективный дефицит IgA, аутоиммунный лимфопролиферативный синдром, аутоиммунный полиýндокринный синдром 1-го типа, синдром Чедиака – Хигаши, дефицит CD70, синдромы тяжелой врожденной нейтропении, дефициты адгезии лейкоцитов, локализованный агрессивный пародонтит, синдром Папийона – Лефевра, хронический кожно-слизистый кандидоз, синдром Маршалла, гипер-IgD синдром, синдром Айкарди – Гутьереса 7-го типа, синдром херувизма, синдром CANDLE (хронический атипичный нейтрофильный дерматит с липодистрофией), PAPA (пиогенный артрит, гангренозная пиодермия и акне), хронический рецидивирующий мультифокальном остеомиелит, периодонтальный синдром Элерса – Данло, дефицит С1-ингибитора. Приведены данные о роли секреторных иммуноглобулинов, определяемых в слюнной жидкости.
Ключевые слова
Об авторах
620049, г. Екатеринбург, ул. Первомайская, 106
Институт иммунологии и физиологии Уральского отделения РАН (ИИФ УрО РАН); Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Россия
канд. мед. наук, ст. науч. сотрудник, 620049, г. Екатеринбург, ул. Первомайская, 106;
620002, г. Екатеринбург, ул. Мира, 19
Институт иммунологии и физиологии Уральского отделения РАН (ИИФ УрО РАН); Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Россия
д-р мед. наук, заслуженный деятель науки Российской Федерации, профессор, гл. науч. сотрудник, 620049, г. Екатеринбург, ул. Первомайская, 106;
620002, г. Екатеринбург, ул. Мира, 19
Институт иммунологии и физиологии Уральского отделения РАН (ИИФ УрО РАН); Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Россия
канд. мед. наук., науч. сотрудник, 620049, г. Екатеринбург, ул. Первомайская, 106;
620002, г. Екатеринбург, ул. Мира, 19
620049, г. Екатеринбург, ул. Первомайская, 106
Список литературы
1. Szczawinska-Poplonyk A. et al. Oral manifestations of primary immune deficiencies in children Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2009; 108 (3): e9–20. DOI: 10.1016/j.tripleo.2009.03.049.
2. Peacock M., Arce R., Cutler C. Periodontal and other oral manifestations of immunodeficiency diseases. Oral Dis. 2016; 23 (7): 866–888. DOI: 10.1111/odi.12584.
3. O’Connell A.C. et al. Delayed eruption of permanent teeth in hyperimmunoglobulinemia E recurrent infection syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2000; 89 (2): 177–185.
4. Domingo D.L. et al. Novel intraoral phenotypes in hyperimmunoglobulin-E syndrome. Oral Dis. 2008; 14 (1): 73–81. DOI: 10.1111/j.1601-0825.2007.01363.x.
5. Freeman A.F., Domingo D.L., Holland S.M. Hyper IgE (Job’s) syndrome: a primary immune deficiency with oral manifestations. Oral Dis. 2009; 15 (1): 2–7. DOI: 10.1111/j.1601-0825.2008.01463.x.
6. Esposito L. et al. Hyper-IgE syndrome: dental implications. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012; 114 (2): 147–153. DOI: 10.1016/j.oooo.2012.04.005.
7. Reddy S.S., Binnal A. Wiscott Aldrich syndrome with oral involvement: A case report. J. Dent. Child. 2011; 78 (1): 49–52. 8. Toka O. et al. Dental aspects in patients with DiGeorge syndrome. Quintessence Int. Berl. Ger. 1985. 2010; 41 (7): 551–556.
8. Nordgarden H. et al. Dental developmental disturbances in 50 individuals with the 22q11.2 deletion syndrome; relation to medical conditions? Acta Odontol. Scand. 2012. 70 (3): 194–201. DOI: 10.3109/00016357.2011.629624.
9. Kulan P., Pekiner F.N., Akyüz S. Oral manifestation and dental management of catch 22 syndrome. Marmara Dental Journal. 2013; 1 (1): 46–48.
10. Sullivan K.E. et al. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin. Immunol. Immunopathol. 1998; 86 (2): 141–146.
11. Gordon S.M., Dionne R.A., Snyder J. Dental fear and anxiety as a barrier to accessing oral health care among patients with special health care needs. Spec. Care Dent. 1998; 18 (2): 88–92.
12. Matevosyan N.R. Oral health of adults with serious mental illnesses: a review. Community Ment. Health J. 2010; 46 (6): 553–562. DOI: 10.1007/s10597-009-9280-x.
13. Klingberg G., Hallberg U., Oskarsdóttir S. Oral health and 22q11 deletion syndrome: thoughts and experiences from the parents’ perspectives. Int. J. Paediatr. Dent. 2010; 20 (4): 283–292. DOI: 10.1111/j.1365-263X.2010.01052.x.
14. Долгих М.А. и др. Распространенность кариеса у детей с ошибками иммунитета. Журнал теоретической и клинической медицины. 2018; 4: 56–58.
15. Feske S., Picard C., Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin. Immunol. Orlando Fla. 2010; 135 (2): 169–182. DOI: 10.1016/j.clim.2010.01.011.
16. Picard C., Casanova J.-L., Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin. Microbiol. Rev. 2011; 24 (3): 490–497. DOI: 10.1128/CMR.00001-11.
17. Meighani G. et al. Oral and dental health status in patients with primary antibody deficiencies. Iran. J. Allergy Asthma Immunol. 2011; 10 (4): 289–293. DOI: 010.04/ ijaai.289293.
18. Fernandes K.S. et al. Salivary immunoglobulins in individuals with common variable immunodeficiency. Braz. Dent. J. 2016; 27 (6): 641–645. DOI: 10.1590/0103- 6440201601096.
19. Yel L. Selective IgA deficiency. J. Clin. Immunol. 2010; 30 (1): 10–16. DOI: 10.1007/s10875-009-9357-x.
20. Thieffry S. et al. Ataxiatelangiectasis (7 personal cases). Rev. Neurol. (Paris). 1961; 105: 390–405.
21. Lakhanpal S. et al. Evidence for linkage of IgA deficiency with the major histocompatibility complex. Mayo Clin. Proc. 1988; 63 (5): 461–465.
22. Wang N., Hammarström L. IgA deficiency: what is new? Curr. Opin. Allergy Clin. Immunol. 2012; 12 (6): 602– 608. DOI: 10.1097/ACI.0b013e3283594219.
23. Yazdani R. et al. Clinical phenotype classification for selective immunoglobulin A deficiency. Expert Rev. Clin. Immunol. 2015; 11 (11): 1245–1254. DOI: 0.1586/1744666X.2015.1081565.
24. Norhagen E.G. et al. Immunoglobulin levels in saliva in individuals with selective IgA deficiency: Compensatory IgM secretion and its correlation with HLA and susceptibility to infections. J. Clin. Immunol. 1989; 9 (4): 279–286.
25. Kiykim A. et al. Comparison of oral microflora in selective IgA deficiency and X linked agammaglobulinemia cases with control group. Turk Pediatri Arsivi. 2013; 48: 204–209. DOI: 10.4274/tpa.438.
26. Azzi L. et al. Oral manifestations of selective IgA-deficiency: review and case-report. J. Biol. Regul. Homeost. Agents. 2017; 31 (2. Suppl. 1): 113–117.
27. Tar I. et al. Oral and dental conditions of children with selective IgA deficiency. Pediatr. Allergy Immunol. Off. Publ. Eur. Soc. Pediatr. Allergy Immunol. 2008; 19: 33–36.
28. Engström G.N. et al. Oral conditions in individuals with selective immunoglobulin A deficiency and common variable immunodeficiency. J. Periodontol. 1992; 63 (12): 984–989.
29. Nikfarjam J. et al. Oral manifestations in selective IgA deficiency. Int. J. Dent. Hyg. 2004; 2 (1): 19–25.
30. Van Nieuw Amerongen A., Bolscher J.G.M., Veerman E.C.I. Salivary proteins: protective and diagnostic value in cariology? Caries Res. 2004; 38 (3): 247–253.
31. Pac M. et al. Recurrent oral inflammation in autoimmune lymphoproliferative syndrome. J. Pediatr. Sci. 2014; 6: е 211. DOI: 10.17334/jps.49665.
32. McGovern E. et al. Oral health in autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED). Eur. Arch. Paediatr. Dent. Off. J. Eur. Acad. Paediatr. Dent. 2008; 9 (4): 236–244.
33. Ahonen P. et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990; 322 (26): 1829–1836.
34. Thumbigere Math V. et al. Periodontitis in Chédiak– Higashi syndrome: An altered immunoinflammatory response. JDR Clin. Transl. Res. 2018; 3 (1): 35–46. DOI: 10.1177/2380084417724117.
35. Khocht A. et al. Periodontitis associated with Chédiak– Higashi syndrome in a young African American male. J. Int. Acad. Periodontol. 2010; 12 (2): 49–55.
36. Delcourt-Debruyne E.M., Boutigny H.R., Hildebrand H.F. Features of severe periodontal disease in a teenager with Chédiak–Higashi syndrome. J. Periodontol. 2000; 71 (5): 816–824.
37. Bailleul-Forestier I. et al. Generalized periodontitis associated with Chédiak – Higashi syndrome. J. Periodontol. 2008; 79 (7): 1263–1270. DOI: 10.1902/jop.2008.070440.
38. Caorsi R. et al. CD70 deficiency due to a novel mutation in a patient with severe Chronic EBV Infection Presenting As a Periodic Fever. Front. Immunol. 2018; 8. DOI: 10.3389/fimmu.2017.02015.
39. Ye Y. et al. Mutations in the ELANE gene are associated with development of periodontitis in patients with severe congenital neutropenia. J. Clin. Immunol. 2011; 31 (6): 936–945. DOI: 10.1007/s10875-011-9572-0.
40. Roberts M., Atkinson J. Oral manifestations associated with leukocyte adhesion deficiency: a five-year case study. Pediatr. Dent. 1990; 12: 107–111.
41. Etzioni A. Leukocyte adhesion deficiency (LAD) syndromes. Orphanet Encycl. 2005; (5): 1–4.
42. Dababneh R. et al. Periodontal manifestation of leukocyte adhesion deficiency type I. J. Periodontol. 2008; 79 (4): 764–768. DOI: 10.1902/jop.2008.070323.
43. Hajishengallis G., Moutsopoulos N.M. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: not a raging infection but a raging inflammatory response. Expert Rev. Clin. Immunol. 2014; 10 (8): 973–975. DOI: 10.1586/1744666X.2014.929944.
44. Zhang Y. et al. Evaluation of human leukocyte N-formylpeptide receptor (FPR1) SNPs in aggressive periodontitis patients. Genes Immun. 2003; 4 (1): 22–29.
45. Khan F.Y., Jan S.M., Mushtaq M. Papillon – Lefèvre syndrome: Case report and review of the literature. J. Indian Soc. Periodontol. 2012; 16 (2): 261–265. DOI: 10.4103/0972-124X.99273.
46. Sharma A., Kaur G., Sharma A. Papillon – Lefevre syndrome: A case report of 2 affected siblings. J. Indian Soc. Periodontol. 2013; 17 (3): 373–377. DOI: 10.4103/0972-124X.115643.
47. Sreeramulu B. et al. Papillon – Lefèvre syndrome: clinical presentation and management options. Clin. Cosmet. Investig. Dent. 2015; 7: 75–81. DOI: 10.2147/CCIDE.S76080.
48. Roberts H. et al. Characterization of neutrophil function in Papillon – Lefèvre syndrome. J. Leukoc. Biol. 2016; 100 (2): 433–444. DOI: 10.1189/jlb.5A1015-489R.
49. Oveisi M., Barzilay O., Hanafi A. Periodontal disease in immunodeficient patients: Clinical guidelines for diagnosis and management. Int. Dent. J. Stud. Res. 2015; 3 (2): 93–104.
50. Wang X., van de Veerdonk F.L. When the fight against fungi goes wrong. PLоS Pathog. 2016; 12 (2): e1005400. DOI: 10.1371/journal.ppat.1005400.
51. Lanternier F. et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J. Allergy Clin. Immunol. 2015; 135 (6): 1558–1568. DOI: 10.1016/j.jaci.2014.12.1930.
52. Glocker E.-O. et al. A Homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009; 361 (18): 1727–1735. DOI: 10.1056/NEJMoa0810719.
54. Okada S. et al. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016; 5 (12): e114. DOI: 10.1038/cti.2016.71.
55. Okada S. et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015; 349 (6248): 606–613. DOI: 10.1126/science.aaa4282.
56. Kraszewska-Głomba B., Matkowska-Kocjan A., Szenborn L. The pathogenesis of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome: A Review of Current Research. 2015; 5: 563876. DOI: 10.1155/2015/563876.
57. Pascual V. et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005; 201 (9): 1479–1486.
58. Cattalini M. et al. Basic characteristics of adults with periodic fever, aphthous stomatitis, pharyngitis, and adenopathy Syndrome in Comparison with the typical pediatric expression of Disease. Mediators Inflamт. 2015; 2015: 11. DOI: 10.1155/2015/570418.
59. Cantarini L. et al. Diagnostic criteria for adult-onset periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome. Front. Immunol. 2017; 8. DOI: 10.3389/fimmu.2017.01018.
60. Berkun Y. et al. The familial mediterranean fever gene as a modifier of periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome. Semin. Arthritis Rheum. 2011; 40 (5): 467–472. DOI: 10.1016/j.semarthrit.2010.06.009.
61. Colotto M. et al. PFAPA Syndrome in a young adult with a history of tonsillectomy. Intern. Med. 2011; 50 (3): 223–225.
62. Bader-Meunier B. et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. 2014; 128 (1): e152– 159. DOI: 10.1542/peds.2010-3639.
63. Drenth J.P., Haagsma C.J., van der Meer J.W. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Baltimore). 1994; 73 (3): 133–144.
64. Rutsch F. et al. A Specific IFIH1 gain-of-function mutation causes singleton – Merten syndrome. Am. J. Hum. Genet. 2015; 96 (2): 275–282. DOI: 10.1016/j.ajhg.2014.12.014.
65. Papadaki M.E. et al. Cherubism: best clinical practice. Orphanet J. Rare Dis. 2012; 7 (Suppl. 1): 6. DOI: 10.1186/1750-1172-7-S1-S6.
66. Meng X.-M., Yu S.-F., Yu G.-Y. Clinicopathologic study of 24 cases of cherubism. Int. J. Oral Maxillofac. Surg. 2005; 34 (4): 350–356.
67. Niranjan B. et al. Non-hereditary cherubism. J. Oral Maxillofac. Pathol. JOMFP. 2014; 18 (1): 84–88. DOI: 10.4103/0973-029X.131920.
68. Carvalho Silva E., Carvalho Silva G.C., Vieira T.C. Cherubism: clinicoradiographic features, treatment, and long-term follow-up of 8 cases. J. Oral Maxillofac. Surg. Off. J. Am. Assoc. Oral Maxillofac. Surg. 2007; 65 (3): 517–522.
69. Roberts T. et al. Candle syndrome: Orodfacial manifestations and dental implications. Head Face Med. 2015; 11: 38.
70. Tallon B., Corkill M. Peculiarities of PAPA syndrome. Rheumatol. Oxf. Engl. 2006; 45 (9): 1140–1143.
71. Smith E.J. et al. Clinical, molecular, and genetic characteristics of PAPA Syndrome: A review. Curr. Genomics. 2010; 11 (7): 519–527. DOI: 10.2174/138920210793175921.
72. Wargo J.J., Emmer B.T. Systemic Inflammation Gone Awry: PASH Syndrome and Temporomandibular Joint Ankylosis. Am. J. Med. 2016; 129 (4): e1–3. DOI: 10.1016/j.amjmed.2015.12.019.
73. Jansson A. et al. Classification of Non-Bacterial OsteitisRetrospective Study of clinical, immunological and genetic aspects in 89 patients. Rheumatology. 2007; 46 (1): 154–160.
74. Monsour P.A.J., Dalton J.B. Chronic recurrent multifocal osteomyelitis involving the mandible: case reports and review of the literature. Dentomaxillofacial Radiol. 2010; 39 (3): 184–190. DOI: 10.1259/dmfr/23060413.
76. Padwa B.L. et al. Pediatric chronic nonbacterial osteomyelitis of the jaw: clinical, radiographic, and histopathologic features. J. Oral Maxillofac. Surg. 2016; 74 (12): 2393–2402. DOI: 10.1016/j.joms.2016.05.021.
77. Glocker E.-O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 Receptor. N. Engl. J. Med. 2009; 361 (21): 2033–2045. DOI: 10.1056/NEJMoa0907206.
78. Kotlarz D. et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012; 143 (2): 347–355. DOI: 10.1053/j.gastro.2012.04.045. 80. Stojanov S., Mcdermott M.F. The tumour necrosis factor receptor-associated periodic syndrome: current concepts. Expert Rev. Mol. Med. 2005; 7 (22): 1–18.
79. Lachmann H.J. et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/ EUROTRAPS international registry. Ann. Rheum. Dis. 2014; 73 (12): 2160–2167. DOI: 10.1136/annrheumdis-2013-204184.
80. Medrano San Ildefonso M., Bruscas Izu C. Hypergammaglobulinemia D syndrome. An. Med. Interna Madr. Spain. 1984. 2000; 17 (4): 213–216.
81. Stewart R.E., Hollister D.W., Rimoin D.L. A new variant of Ehlers-Danlos syndrome: an autosomal dominant disorder of fragile skin, abnormal scarring, and generalized periodontitis. Birth Defects Orig. Artic. Ser. 1977; 13 (3B): 85–93.
82. Kapferer-Seebacher I. et al. Periodontal Ehlers-Danlos Syndrome Is Caused by Mutations in C1R and C1S, which Encode Subcomponents C1r and C1s of Complement. Am. J. Hum. Genet. 2016; 99 (5): 1005–1014. DOI: 10.1016/j.ajhg.2016.08.019.
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Селективный IgA дефицит
Селективный дефицит IgA диагностируют при уровне IgA 7 мг/дл ( 70 мг/л 0,4375 мкмоль/л) с нормальными уровнями IgG и IgM. Это наиболее частый первичный иммунодефицит Первичные иммунодефициты Иммунодефицитные состояния связаны или предрасполагают пациентов к осложнениям различного рода, в том числе к инфекциям, аутоиммунным нарушениям, лимфомам и другим онкозаболеваниям. Первичные. Прочитайте дополнительные сведения . У многих пациентов он протекает бессимптомно, но у других развиваются рецидивирующие инфекции и аутоиммунные процессы. У некоторых пациентов со временем развивается вариабельный неклассифицируемый иммунодефицит Общий вариабельный иммунодефицит (ОВИ) Общий вариабельный иммунодефицит (приобретенная гипогаммаглобулинемия или гипогаммаглобулинемия с поздним началом) характеризуется низким уровнем lg с фенотипически нормальными В-лимфоцитами. Прочитайте дополнительные сведения , у других отмечается спонтанное улучшение. Диагноз ставят по результатам измерения сывороточных иммуноглобулинов. При необходимости, лечение антибиотиками (иногда профилактическое) и обычно отказ от препаратов крови, содержащих IgA.
Схема наследования неизвестна, но наличие у члена семьи селективного дефицита IgA увеличивает риск заболевания примерно в 50 раз.
У некоторых пациентов присутствуют мутации гена TACI (трансмембранный активатор и партнёр кальциевого модулятора и лиганда циклофилина). Селективный дефицит IgA также обычно встречается у пациентов с определенными HLA-гаплотипами; часто присутствуют редкие аллели или делеции генов главного комплекса гистосовместимости Лейкоцитарные антигены человека (ЛАЧ) (МНС) региона класса III.
Симптомы и признаки селективного дефицита IgA
У многих пациентов с изолированной недостаточностью IgA-типа заболевание протекает бессимптомно; у других отмечаются рецидивирующие синусно-пульмональные инфекции, диарея, аллергические состояния (такие, как астма, сопровождающаяся носовыми полипами), или аутоиммунные нарушения (такие, как целиакия или воспалительные заболевания кишечника, системная красная волчанка, хронический активный гепатит).
Анти-IgA антитела могут вырабатываться после воздействия IgA при трансфузии, иммуноглобулина (Ig) или других препаратов крови; в редких случаях, если при повторном введении этих препаратов у пациента может развиться анафилактическая реакция.
Диагностика изолированной недостаточности IgA
Измерение уровней сывороточного Ig
Измерение выработки антител в ответ на вакцинные антигены
Диагноз изолированная недостаточность IgA подозревается у пациентов с рецидивирующими инфекциями (в том числе лямблиозом), анафилактическими реакциями на трансфузию или при наличии в семейном анамнезе вариабельного неклассифицируемого иммунодефицита Общий вариабельный иммунодефицит (ОВИ) Общий вариабельный иммунодефицит (приобретенная гипогаммаглобулинемия или гипогаммаглобулинемия с поздним началом) характеризуется низким уровнем lg с фенотипически нормальными В-лимфоцитами. Прочитайте дополнительные сведения (ВНИ), дефицита IgA или аутоиммунных нарушений, или также у пациентов, принимавших препараты, которые могут вызвать дефицит IgA.
Подтверждение диагноза осуществляется при наличии уровня сывороточного IgA 7 мг/дл (
Поскольку большинство пациентов с низким IgA не имеют клинически значимых проявлений, исследование членов семьи не рекомендуется.
Прогноз при селективном дефиците ИгА (IgA)
У небольшого количества пациентов с недостаточностью IgA развивается ОВИД; у других отмечается спонтанное улучшение. Прогноз ухудшается при развитии аутоиммунных процессов.
Лечение селективного IgA-дефицита
Прием антибиотиков необходим как для лечения, так и для профилактики в тяжелых случаях
Исключение контактов с продуктами крови, содержащими IgA
Лечение аллергических проявлений. Антибиотики назначают при необходимости лечения бактериальных инфекций уха, придаточных пазух носа, легких, желудочно-кишечного и мочеполового трактов и, в тяжелых случаях, профилактически.
Следует избегать применения препаратов крови, содержащих IgA, поскольку IgA приводит к развитию анти-IgА-опосредованной анафилактической реакции. При необходимости проведения трансфузии эритроцитарной массы используют только отмытые упакованные эритроциты. Большинство заместителей иммуноглобулинов, представленных сегодня на рынке, имеют минимальное содержание IgA и не приводят к нежелательным явлениям даже у пациентов с дефицитом IgA.
Поскольку заместительная терапия иммуноглобулинами содержит главным образом IgG, она не оказывает благоприятного воздействия на пациентов с дефицитом IgA, также возможен риск анафилактических реакций по причине того, что у пациентов могут вырабатываться анти-IgA антитела. В редких случаях, если у пациентов не наблюдалась продукция антител в ответ на вакцинацию, и если профилактические антибиотики были неэффективны, могут быть полезны специально разработанные иммуноглобулиновые препараты, содержащие предельно низкие уровни IgA. При необходимости других компонентов крови, они должны быть IgA-дефицитными и вымытыми.
Пациентам с изолированной недостаточностью IgA-типа рекомендовано носить браслет идентификации, во избежание случайного введения плазмы или иммуноглобулинов, что может привести к анафилактическому шоку.
Основные положения
Селективный дефицит IgA является наиболее частым первичным иммунодефицитом.
Заболевание у пациентов может протекать бессимптомно или с развитием рецидивирующих инфекций или аутоиммунных нарушений; у некоторых пациентов со временем развивается ВНИ, но у других селективный дефицит IgA разрешается спонтанно.
Селективный дефицит IgA подозревается в случае, если у пациентов развиваются анафилактические реакции на трансфузию, прием лекарственных препаратов, приводящих к дефициту IgA, а также наблюдаются рецидивирующие инфекции или соответствующий семейный анамнез.
Подтверждение диагноза осуществляют путем измерения уровней Ig и титров антител после введения вакцины; диагностическими являются уровни IgA
Антибиотики применяют при необходимости, а в тяжелых случаях - в профилактических целях.
Следует избегать применения для пациентов препаратов крови ли иммунных глобулинов, которые содержат более чем минимальное количество IgA.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Дефицит иммуноглобулина А
Дефицит иммуноглобулина А – группа первичных иммунодефицитных состояний, которые обусловлены нарушением синтеза или ускоренным разрушением молекул иммуноглобулинов данного класса. Симптомами заболевания являются частые бактериальные инфекции (особенно респираторной системы и ЛОР-органов), нарушения со стороны желудочно-кишечного тракта, аллергии и аутоиммунные поражения. Диагностика дефицита иммуноглобулина А производится путем определения его количества в сыворотке крови, также применяют молекулярно-генетические техники. Лечение симптоматическое, сводится к профилактике и своевременной терапии бактериальных инфекций и других нарушений. В некоторых случаях осуществляют заместительную иммуноглобулиновую терапию.
Общие сведения
Дефицит иммуноглобулина А – полиэтиологическая форма первичного иммунодефицита, при которой наблюдается недостаток этого класса иммуноглобулинов при нормальном содержании остальных классов (G, M). Дефицит может быть полным, с резким снижением всех фракций глобулина А, и селективным, с недостатком только определенных подклассов этих молекул. Селективный дефицит иммуноглобулина А является очень распространенным состоянием, по некоторым данным его встречаемость составляет 1:400-600. Явления иммунодефицита при селективном недостатке соединения достаточно стертые, почти у двух третей больных заболевание не диагностируется, поскольку они не обращаются за медицинской помощью. Врачи-иммунологи установили, что дефицит иммуноглобулина А может проявляться не только инфекционными симптомами, у пациентов также нередко наблюдаются обменные и аутоиммунные расстройства. С учетом этого обстоятельства можно предположить, что встречаемость данного состояния еще выше, чем предполагалось ранее. Современные генетики считают, что заболевание возникает спорадически или является наследственной патологией, причем в качестве механизма передачи может выступать как аутосомно-доминантный, так и аутосомно-рецессивный путь наследования.
Причины дефицита иммуноглобулина А
Этиология и патогенез как полного, так и селективного дефицитов иммуноглобулина А на сегодняшний момент до конца не определены. Пока удалось установить лишь генетически-молекулярные механизмы отдельных форм заболевания. Например, селективный дефицит иммуноглобулина А типа 2 обусловлен мутациями гена NFRSF13B, локализованного на 17-й хромосоме и кодирующего одноименный белок. Данный протеин представляет собой трансмембранный рецептор на поверхности В-лимфоцитов, отвечает за распознавание фактора некроза опухолей и некоторых других иммунокомпетентных молекул. Соединение принимает активное участие в регуляции интенсивности иммунного ответа и секреции различных классов иммуноглобулинов. По данным молекулярных исследований, генетический дефект гена TNFRSF13B, приводящий к развитию аномального рецептора, делает определенные фракции В-лимфоцитов функционально незрелыми. Такие клетки вместо продукции оптимальных количеств иммуноглобулинов А выделяют смесь из классов А и D, что приводит к уменьшению концентрации класса А.
Иммуноглобулин А отличается от других родственных молекул тем, что обуславливает самый первый этап неспецифической иммунологической защиты организма, поскольку выделяется в составе секрета желез слизистых оболочек. При его недостатке патогенным микроорганизмам становится легче внедряться в слабо защищенные нежные ткани слизистых дыхательных путей, желудочно-кишечного тракта и ЛОР-органов. Механизмы аутоиммунных, обменных и аллергических нарушений при дефиците иммуноглобулина А до сих пор неизвестны. Существует предположение, что его низкая концентрация вносит дисбаланс во всю иммунную систему.
Симптомы дефицита иммуноглобулина А
Все проявления дефицита иммуноглобулина А в иммунологии разделяют на инфекционные, обменные (или желудочно-кишечные), аутоиммунные и аллергические. Инфекционные симптомы заключаются в повышенной частоте бактериальных инфекций дыхательных путей – у больных часто возникают ларингиты, трахеиты, бронхиты и пневмонии, которые могут принимать тяжелое течение и сопровождаться развитием осложнений. Кроме того, для дефицита иммуноглобулина А характерен быстрый переход острых воспалительных процессов в хронические формы, что особенно показательно в отношении поражений ЛОР-органов – у пациентов нередко диагностируются отиты, гаймориты и фронтиты. Достаточно часто встречающийся сочетанный дефицит иммуноглобулинов А и G2 приводит к тяжелым обструктивным поражениям легких.
В меньшей степени инфекционные поражения затрагивают желудочно-кишечный тракт. При дефиците иммуноглобулина А наблюдается некоторое учащение лямблиоза, могут регистрироваться гастриты и энтериты. Наиболее характерными для этого иммунодефицита симптомами со стороны ЖКТ являются непереносимость лактозы и целиакия (невосприимчивость белка злаковых глютена), которые при отсутствии коррекции питания могут привести к атрофии кишечных ворсин и синдрому мальабсорбции. Среди больных дефицитом иммуноглобулина А также часто регистрируются язвенный колит, билиарный цирроз печени и хронические гепатиты аутоиммунного генеза. Перечисленные заболевания сопровождаются болями в животе, частыми эпизодами диареи, похуданием и гиповитаминозами (по причине нарушения всасывания нутриентов из-за мальабсорбции).
Помимо вышеописанных заболеваний желудочно-кишечного тракта, аутоиммунные и аллергические поражения при дефиците иммуноглобулина А проявляются повышенной частотой развития системной красной волчанки и ревматоидного артрита. Возможны также тромбоцитопеническая пурпура и аутоиммунная гемолитическая анемия, нередко – с тяжелым течением. Более чем у половины больных в крови определяются аутоантитела против собственного иммуноглобулина А, что еще более усугубляет явления недостатка данного соединения. У пациентов с дефицитом иммуноглобулина А часто выявляют крапивницу, атопические дерматиты, бронхиальную астму и другие заболевания аллергического происхождения.
Диагностика дефицита иммуноглобулина А
Диагностика дефицита иммуноглобулина А производится на основании данных истории болезни пациента (частые инфекции дыхательных путей и ЛОР-органов, поражения ЖКТ), но наиболее точным способом подтверждения диагноза является определение количества сывороточных иммуноглобулинов разных классов. При этом может обнаруживаться изолированное уменьшение уровня этого компонента гуморального иммунитета ниже 0,05 г/л, что свидетельствует о его дефиците. На этом фоне уровень иммуноглобулинов G и M остается в пределах нормы, иногда выявляется снижение фракции G2. При частичном дефиците иммуноглобулина А его концентрация остается в пределах 0,05-0,2 г/л. При оценке результатов анализа важно помнить о возрастных особенностях количества глобулинов в плазме крови – например, концентрация фракции А 0,05-0,3 г/л у детей до 5-ти лет носит название транзиторного дефицита и может исчезать в дальнейшем.
Иногда обнаруживается парциальный дефицит иммуноглобулина А, при котором его количество в плазме снижено, но концентрация соединения в выделениях слизистых оболочек достаточно высока. Никаких клинических симптомов заболевания у пациентов с парциальным дефицитом не выявляется. В иммунограмме следует обратить внимание на количество и функциональную активность иммунокомпетентных клеток. При дефиците иммуноглобулина А количество Т- и В-лимфоцитов обычно сохранено на нормальном уровне, снижение количества Т-лимфоцитов свидетельствует о возможном наличии общего вариабельного иммунодефицита. Среди других методов диагностики вспомогательную роль играют определение в плазме антинуклеарных и других аутоантител, автоматическое секвенирование гена TNFRSF13B и аллергологические пробы.
Лечение, прогноз и профилактика дефицита иммуноглобулина А
Специфическое лечение данного иммунодефицита отсутствует, в некоторых случаях производят заместительную иммуноглобулиновую терапию. В основном используют антибиотики для лечения бактериальных инфекций, иногда назначают профилактические курсы антибактериальных средств. Необходима коррекция рациона питания (исключение опасных продуктов) при развитии пищевой аллергии и целиакии. В последнем случае исключают блюда на основе злаков. Бронхиальную астму и другие аллергические патологии лечат общепринятыми препаратами – антигистаминными и бронхолитическими средствами. При выраженных аутоиммунных нарушениях назначают иммуносупрессивные препараты – кортикостероиды и цитостатики.
Прогноз при дефиците иммуноглобулина А в целом благоприятный. У многих больных патология протекает абсолютно бессимптомно и не требует специального лечения. При повышении частоты бактериальных инфекций, аутоиммунных поражениях и нарушениях всасывания (синдроме мальабсорбции) прогноз может ухудшаться соответственно тяжести симптомов. Для профилактики развития перечисленных проявлений необходимо использование антибиотиков при первых признаках инфекционного процесса, соблюдение правил относительно режима питания и состава рациона, регулярное наблюдение у иммунолога и врачей других специальностей (в зависимости от сопутствующих нарушений). Следует соблюдать осторожность при переливании цельной крови или ее компонентов – в редких случаях у больных наблюдается анафилактическая реакция из-за наличия в крови аутоантител к иммуноглобулину А.
Избирательный дефицит IgA – один из самых частых случаев иммунодефицита
В крови лиц, страдающих этим заболеванием, уровень содержания иммуноглобулина A снижен, или белок вообще отсутствует.
Причины
Как правило, дефицит IgA является наследственным, то есть передается детям от родителей. Однако в некоторых случаях дефицит IgA может быть связан с приемом лекарственных препаратов.
Частота встречаемости заболевания среди представителей европеоидной расы составляет 1 случай на 700 человек. Среди представителей других рас частота встречаемости ниже.
Симптомы
В большинстве случаев избирательный дефицит IgA протекает бессимптомно.
Среди симптомов заболевания можно назвать частые эпизоды:
• Бронхита
• Диареи
• Конъюнктивита (инфекционное заболевание глаз)
• Инфекций полости рта
• Среднего отита (инфекционное заболевание среднего уха)
• Пневмонии
• Синусита
• Инфекций кожи
• Инфекций верхних дыхательных путей.
К другим симптомам относятся:
• Бронхоэктатическая болезнь (заболевание, при котором происходит расширение участков бронхов)
• Бронхиальная астма неизвестного происхождения.
Диагностика
Для дефицита IgA характерен семейный анамнез. Установить диагноз позволяют определенные показатели:
• IgA
• IgG
• Подклассов IgG
• IgM
и методы исследования:
• Определение количества иммуноглобулинов
• Иммуноэлектрофорез белков сыворотки крови.
Лечение
Специфическое лечение не разработано. В некоторых случаях уровень содержания IgA самостоятельно восстанавливается до нормальных значений.
Для лечения инфекционных заболеваний используются антибиотики. С целью профилактики рецидива некоторым пациентам назначают длительные курсы антибиотиков.
Если избирательный дефицит IgA сопровождается дефицитом подклассов IgG, пациентам внутривенно вводят иммуноглобулины.
Примечание: внутривенное введение препаратов крови и иммуноглобулинов при отсутствии IgA ведет к выработке антител к IgA. У пациентов развиваются аллергические реакции, вплоть до анафилактического шока, представляющего угрозу для жизни. Таким пациентам IgA вводить нельзя.
Прогноз
Селективный дефицит IgA менее опасен, чем другие иммунодефициты. У некоторых пациентов уровень содержания IgA постепенно нормализуется, и происходит спонтанное выздоровление.
Возможные осложнения
На фоне селективного дефицита IgA могут развиться аутоиммунные заболевания (ревматоидный артрит, системная красная волчанка) или целиакия.
В ответ на введение препаратов в крови у пациентов с дефицитом IgA могут вырабатываться антитела к IgA, что сопровождается тяжелыми аллергическими реакциями. Если пациенту требуется переливание крови, ему следует вводить отмытые клетки.
В каком случае следует обратиться к врачу
Если у ближайших родственников пары, планирующей завести ребенка, были случаи селективного дефицита IgA, будущим родителям требуется генетическое консультирование.
Если врач планирует вводить пациенту иммуноглобулины или препараты крови, пациент должен предупредить врача о том, что у него дефицит IgA.
Профилактика
Профилактика селективного дефицита IgA заключается в генетическом консультировании будущих родителей, имеющих семейный анамнез данного заболевания.
Клинические особенности и опыт ведения больных первичными иммунодефицитами с нарушением антителопродукции в Свердловской области
Авторы: Бельтюков Е.К. 1 , Скороходов И.С. 2 , Виноградов А.В. 3 , Наумова В.В. 1
1 ФГБОУ ВО УГМУ Минздрава России, Екатеринбург, Россия
2 Многопрофильная клиника «Ставко», Екатеринбург
3 Министерство здравоохранения Свердловской области, Екатеринбург
Цель исследования: выявить клинические особенности и оценить эффективность заместительной терапии стандартными внутривенными иммуноглобулинами (ВВИГ) у больных первичными иммунодефицитами (ПИД) с дефектами антителообразования в Свердловской области.
Материал и методы: в исследование включены пациенты регионального регистра ПИД Свердловской области, в котором 81% (n=91) контингента имеет дефекты антителообразования.
Результаты: наиболее распространенным вариантом ПИД в регистре Свердловской области является селективный дефицит иммуноглобулина A (IgА) (n=48). Селективный дефицит IgA сопровождается аллергическими заболеваниями и инфекционными процессами нетяжелого течения, не требующими специальных методов лечения. Другие формы ПИД с дефектами антителообразования (n=37) в регистре Свердловской области представлены общей вариабельной иммунной недостаточностью, агаммаглобулинемией, синдромом гиперпродукции иммуноглобулина IgЕ. Для пациентов с этими формами ПИД характерны тяжелые рецидивирующие инфекционные заболевания. У пациентов с ПИД в Свердловской области чаще всего регистрируются хронический риносинусит (n=18), бронхоэктатическая болезнь (n=18), пневмонии (n=18). Большинство больных с гуморальными видами иммунодефицитов нуждаются в пожизненной заместительной терапии донорскими иммуноглобулинами. У пациентов (n=19), получающих в качестве патогенетического лечения заместительную терапию стандартными ВВИГ, удалось снизить частоту рецидивов и обострений хронического риносинусита в 2,7 раза.
Заключение: ведущими клиническими формами ПИД в региональном регистре Свердловской области выступают дефекты антителообразования: селективный дефицит IgА и общая вариабельная иммунная недостаточность. Клинически ПИД проявляются в основном в форме рецидивирующих, резистентных к терапии инфекционных заболеваний органов дыхания, которые требуют специальных методов лечения. Единственным высокоэффективным методом лечения ПИД с нарушением антителообразования является заместительная терапия стандартными ВВИГ.
Ключевые слова: первичные иммунодефициты, дефекты антителообразования, селективный дефицит иммуноглобулина А, общая вариабельная иммунная недостаточность, Х-сцепленная агаммаглобулинемия, синдром гиперпродукции иммуноглобулина Е, заместительная терапия, внутривенные иммуноглобулины.
Для цитирования: Бельтюков Е.К., Скороходов И.С., Виноградов А.В., Наумова В.В. Клинические особенности и опыт ведения больных первичными иммунодефицитами с нарушением антителопродукции в Свердловской области // РМЖ. 2016. № 16. С. 1108–1111.
Сlinical features and management of patients with primary antibody production deficiencies in Sverdlovsk region
Beltyukov E.K. 1 , Skorokhodov I.S. 2 , Vinogradov A.V. 3 , Naumova V.V. 1
1 Urals State Medical University, Ekaterinburg
2 Stavko Clinic, Ekaterinburg
3 The Ministry of Health of the Sverdlovsk region, Ekaterinburg
Objective: to identify the clinical features and evaluate the effectiveness of replacement therapy with standard intravenous immunoglobulins (IVIGs) in patients with primary antibody production deficiencies in the Sverdlovsk region.
Methods: The study included patients of a regional primary immunodeficiency (PID) register of the Sverdlovsk region, where 81% (n = 91) of patients has defects in antibody production.
Results: Selective IgA deficiency is the most common form of PID in the Sverdlovsk region register (n = 48). Clinical features of selective IgA deficiency are concomitant allergic diseases and infectious processes of mild to moderate course, which do not require special treatment. The other forms of primary antibody production deficiencies (n = 37) in the register of the Sverdlovsk region are submitted to common variable immunodeficiency, agammaglobulinemia, hyper-IgE-syndrome.
Severe recurrent infections are characteristic for patients with these forms of PID. Chronic rhinosinusitis (n = 18), bronchiectasis (n = 18), pneumonia (n 18) are most often detected in PID patients in the Sverdlovsk region. The majority of patients with humoral immunodeficiencies requires lifelong replacement therapy with donor immunoglobulins. The frequency of relapses and exacerbations of chronic rhinosinusitis was 2,7-fold decreased in our patients receiving replacement therapy with standard IVIG as pathogenetic treatment (n = 19).
Conclusion: The prevalent clinical forms of PID in the regional register of Sverdlovsk region are defects in antibody production: selective IgA deficiency and common variable immunodeficiency. The most common clinical manifestations of PID are recurrent, resistant to usual therapy, infectious diseases of respiratory system, which require special treatment. The only and highly effective method of primary antibody production deficiencies treatment is replacement therapy with standard IVIGs.
Key words: primary immunodeficiency, antibody production deficiencies, selective IgA deficiency, common variable immunodeficiency, X-linked agammaglobulinemia, hyper-IgE-syndrome, replacement therapy, intravenous immunoglobulins.
For citation: Beltyukov E.K., Skorokhodov I.S., Vinogradov A.V., Naumova V.V. Сlinical features and management of patients with primary antibody production deficiencies in Sverdlovsk region // RMJ. 2016. № 16. P. 1108–1111.
В статье приведены клинические особенности и опыт ведения больных первичными иммунодефицитами
Введение
Первичные иммунодефициты (ПИД) с нарушением гуморального звена иммунной системы представляют собой генетически детерминированные заболевания, характеризующиеся нарушением процесса антителообразования. ПИД клинически характеризуются развитием инфекционных процессов, склонностью к появлению аутоиммунных заболеваний, злокачественных новообразований. По литературным данным, дефекты с преимущественным поражением гуморального звена иммунитета составляют около 60% от всех ПИД.
Распространенность ПИД с дефектами антителообразования варьирует в зависимости от выявленного дефекта: с селективным дефицитом IgA – 1:300–1:700; общей вариабельной иммунной недостаточностью (ОВИН) – 1:7000–1:200 000; Х-сцепленной агаммаглобулинемией – 1:50 000–1:1 000 000 [1, 2].
Цель исследования: выявить клинические особенности и оценить эффективность заместительной терапии стандартными ВВИГ у больных ПИД с дефектами антителообразования в Свердловской области.
Материал и методы
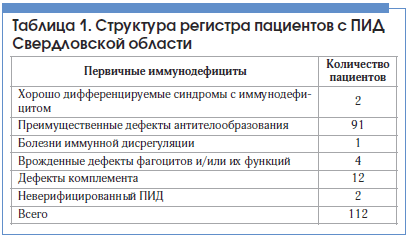
В исследование включены пациенты регионального регистра ПИД Свердловской области, где 81% (n=91) больных имеют дефекты антителообразования. Структура регистра ПИД среди взрослого населения Свердловской области представлена в таблице 1.
Результаты исследования и их обсуждение
Наиболее распространенным вариантом ПИД является селективный дефицит иммуноглобулина А (IgА), при котором уровень сывороточного IgA составляет не более 5 мг/дл при нормальных количественных показателях других звеньев иммунной системы. Клинически селективный дефицит IgA проявляется частыми острыми инфекциями дыхательных путей, желудочно-кишечными заболеваниями инфекционной и неинфекционной природы. Помимо этого, селективному дефициу IgA сопутствует с повышенной частотой весь спектр аллергических заболеваний: аллергический ринит, конъюнктивит, крапивница, атопический дерматит, бронхиальная астма (БА). Как правило, БА у этих пациентов протекает тяжело, что, возможно, связано с частыми респираторными инфекциями, усугубляющими тяжесть БА [1, 2].
Среди аллергических заболеваний, имеющих место у пациентов с селективным дефицитом IgA, учтенных в региональном регистре ПИД Свердловской области (n=48), – атопический дерматит (n=8), бронхиальная астма (n=5), острая крапивница (n=1). Также у этих больных отмечались острые респираторные заболевания (ОРЗ, n=12), хронический тонзиллит (n=6), хронический отит (n=3), хронический гайморит (n=2), хронический бронхит (n=5), в т. ч. в сочетании с аллергическими заболеваниями.
Пациенты с селективным дефицитом IgA не требуют специального патогенетического лечения. Лечебные мероприятия сводятся к терапии вторичных осложнений инфекционной, аллергической или аутоиммунной природы, а также активации сохранных звеньев иммунитета с помощью иммуномодуляторов для компенсации недостаточной продукции IgA.
Другие формы ПИД с дефектами антителообразования (n=37) – ОВИН, агаммаглобулинемия, синдром гиперпродукции иммуноглобулина Е (ГИЕ) – отличаются выраженными клиническими проявлениями в виде хронических заболеваний органов дыхания, кожи, рецидивирующих бронхолегочных инфекций, нередко тяжелых, резистентных к терапии, что требует применения специальных методов лечения. В региональном регистре ПИД Свердловской области у таких пациентов выявлены хронический риносинусит (ХРС, n=18), хронический средний отит (n=6), хронический бронхит (n=3), бронхоэктатическая болезнь (БЭБ, n=18), хроническая обструктивная болезнь легких (n=2), рецидивирующие более 1 раза в год пневмонии (n=18), экзема (n=6). Таким образом, по данным областного регистра ПИД, наиболее часто у больных с ОВИН, агаммаглобулинемией и синдромом ГИЕ развиваются ХРС, БЭБ и пневмонии.
Необходимо отметить, что примерно одну треть областного регистра больных ПИД составляют пациенты с дефектом продукции IgG. В настоящее время единственным, доказавшим высокую эффективность специальным методом лечения данного вида иммунодефицитных состояний является заместительная терапия стандартными ВВИГ.
В рандомизированных исследованиях было показано, что использование ВВИГ в дозе 0,4–0,6 г/кг массы тела в целях поддерживающей заместительной терапии обычно достаточно для достижения желаемого претрансфузионного уровня IgG 6–8 г/л (600–800 мг/дл) [3]. Доза иммуноглобулина должна быть индивидуализирована для каждого пациента с учетом сопутствующей патологии, метаболических особенностей, наличия или отсутствия обострения инфекционного процесса и др. (претрансфузионный уровень является лишь дополнительным параметром оценки эффективности терапии) [2].
Для заместительной терапии ВВИГ используются 5% и 10% стандартные иммуноглобулины, содержащие не менее 95% IgG и прошедшие не менее 3-х стадий вирусной инактивации [4].
Побочные явления при заместительной терапии ВВИГ наблюдаются примерно у 5% пациентов и характеризуются разнообразными симптомами: головной болью, тошнотой, головокружением, болями в животе, диареей, артериальной гипертонией или гипотонией, гипертермией, тахикардией, а в исключительных случаях – тяжелой гипотонией и коллапсом [2].
Применение препаратов 10% иммуноглобулинов не менее чем в 2 раза сокращает продолжительность внутривенной инфузии по сравнению с препаратами 5% ВВИГ, что экономит время медицинских работников и облегчает процедуру для пациентов.
Современный представитель 10% иммуноглобулинов Привиджен обладает также рядом других особенностей, повышающих его эффективность и безопасность и улучшающих общую переносимость инфузий. В частности, Привиджен содержит не менее 98% IgG и самую низкую среди остальных препаратов иммуноглобулинов концентрацию IgA – 0,025 мг/мл. Кроме того, содержащийся в препарате L-пролин в качестве стабилизатора предотвращает агрегацию и фрагментацию IgG. Еще одним достижением производителей данного препарата стала уникальная технология Ig IsoLo®, до предела минимизирующая и без того невысокие риски развития гемолиза и ассоциированных с ним побочных эффектов.
В настоящее время в Свердловской области ВВИГ проводится 19 пациентам с агаммаглобулинемией (n=3), синдромом ГИЕ (n=1), ОВИН (n=15), в т. ч. с использованием Привиджена. Назначение Привиджена пациентке с анафилактическими реакциями на другие стандартные ВВИГ импортного производства позволило продолжить поддерживающую заместительную терапию ВВИГ без каких-либо осложнений, подтвердив высокое качество очистки этого препарата.
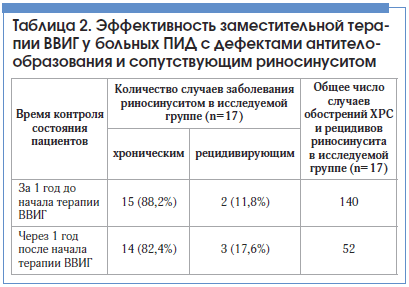
Эффективность ВВИГ оценивалась по динамике течения одной из самых часто встречающихся среди этой группы пациентов патологии – риносинусита. Диагноз ХРС устанавливался с учетом критериев, изложенных в Европейских рекомендациях по риносинуситу (European position paper on rhinosinusitis and nasal polyps – EPOS) 2012 г.: учитывались пациенты, у которых симптомы риносинусита присутствовали более 12 нед. Рецидивирующим считался синусит, с 1–4 эпизодами острой формы заболевания в год, с периодами между обострениями не менее 8 нед., когда симптомы заболевания отсутствуют и лечение не проводится [5]. Обострение ХРС оценивалось согласно критериям Американского общества по инфекционным болезням (IDSA) 2012 г., в соответствии с которыми основными симптомами обострения ХРС являются гнойные выделения из полости носа, гнойные выделения, стекающие по задней стенке глотки, заложенность носа, чувство распирания, давления, боль в области лица, гипосмия или аносмия, лихорадка, а также дополнительные симптомы: головная боль, боль в ухе, неприятный запах изо рта, боль в зубах, кашель, утомляемость.
Диагноз обострения ХРС устанавливался при наличии не менее 2-х основных симптомов или одного основного и не менее чем 2-х дополнительных симптомов [6].
Состояние пациентов, получающих заместительную терапию ВВИГ (n=19), оценивалось за 1 год до начала лечения и по прошествии 1 года лечения. Из 19 пациентов, получающих ВВИГ, диагноз риносинусита был установлен у 17 человек. Применение заместительной терапии ВВИГ у больных ПИД с дефектами антителообразования и сопутствующим риносинуситом привело к уменьшению общего числа случаев обострений ХРС и рецидивов риносинусита в 2,7 раза (табл. 2).
Заключение
В региональном регистре первичных иммунодефицитов Свердловской области преобладают больные ПИД с дефектами антителообразования (81%). Ведущими клиническими формами ПИД являются селективный дефицит IgА и ОВИН. Сопутствующая патология у больных селективным дефицитом IgА представлена в основном атопическим дерматитом, бронхиальной астмой, частыми ОРЗ. ОВИН часто сопровождается бронхоэктатической болезнью, риносинуситом, пневмониями и эффективно лечится в режиме поддерживающей заместительной терапии ВВИГ. Имеющиеся в нашей стране современные ВВИГ 10% концентрации предоставляют врачам достаточную возможность для подбора препарата с учетом индивидуальных особенностей отдельного пациента, тем самым позволяя сделать еще один шаг в сторону персонифицированного подхода к терапии рассматриваемого контингента больных.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Читайте также: