Симптоматическая терапия миелодиспластического синдрома (МДС) - переливание эритроцитов, эритропоэтин
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Открытое исследование фазы 1b R289, ингибитора IRAK1/4, у пациентов с миелодиспластическим синдромом низкого риска (LR MDS), которые рефрактерны/резистентны к предшествующей терапии
Исследование будет открытым, исследование фазы 1b R289 для определения переносимости и предварительная эффективность у пациентов с LR МДС, которые являются рецидивирующими, рефрактерными/резистентными, непереносимость или неадекватная реакция на предшествующую терапию, такую как эритропоэтин (ЭПО), тромбопоэтин (ТПО), луспатерцепт или гипометилирующие агенты (ГМА) при МДС.
Тип вмешательства: Лекарство
Описание: таблетка 250 мг
Этикетка Arm Group: Увеличение дозы
Критерии включения: - Возраст пациента должен быть ≥ 18 лет на момент подписания информированного согласия. - Должен иметь окончательный диагноз МДС с очень низким, низким или промежуточным риском-1. (Международная прогностическая система оценки (IPSS)-R ≤ 3,5) и ≤5% костного мозга миелобласты. - Должен быть рецидивирующим, рефрактерным/резистентным, непереносимым или иметь неадекватную реакцию на все терапии с известными клиническими преимуществами при МДС, такие как TPO, EPO, luspatercept и HMA (например, азацитидин или децитабин). Пациенты с del (5q) должны были ранее не иметь успеха. терапия леналидомидом. - Должен соответствовать хотя бы одному из критериев, связанных с заболеванием, для переливания эритроцитов, тромбоцитов число или абсолютное количество нейтрофилов (АНК) в течение 8 недель до первоначального введения лечение исследования: 1. Симптоматическая анемия без переливания гемоглобина < 9,0 г/дл в течение 8 недель после регистрация или переливание эритроцитов (эритроцитарной массы) определяется как получение >3 единицы эритроцитарной массы (PRBC) за предшествующие 16 недель в течение гемоглобин 60 мл/мин и отсутствие терминальной стадии почечной недостаточности (с использованием Кокрофта-Голта) Критерий исключения: - Предыдущее лечение МДС (т. е. ТПО, ЭПО, луспатерцепт, ГМА) завершилось < 4 недель до начала лечения - Клинически значимая анемия в результате дефицита железа, B12 или фолиевой кислоты, аутоиммунный или наследственный гемолиз или желудочно-кишечное кровотечение. - Документально подтвержденный дефицит железа. Все субъекты должны иметь подтвержденные запасы железа в костном мозге. Если окраска костного мозга железом недоступна, насыщение трансферрина должно быть >20% или ферритин сыворотки > 100 нг/100 мл. - МДС, вторичный по отношению к лечению лучевой терапией, химиотерапией и/или иммунотерапией для злокачественные или аутоиммунные заболевания. - Диагностика хронического миеломоноцитарного лейкоза. - Текущие или предыдущие бласты костного мозга > 5%. - История неконтролируемых судорог. - Неконтролируемая бактериальная или вирусная инфекция (например, подтвержденный ВИЧ, гепатит В или гепатит С). - История активного злокачественного новообразования в течение последних 2 лет до включения в исследование, с исключение из: 1. Адекватно леченный in situ рак шейки матки 2. Адекватно пролеченная базально-клеточная карцинома или локализованная плоскоклеточная карцинома кожа, или 3. Любое другое злокачественное новообразование с ожидаемой продолжительностью жизни более 2 лет. - История или активное, клинически значимое, сердечно-сосудистое, респираторное, желудочно-кишечное, почечное, печеночная, неврологическая, психиатрическая, костно-мышечная, мочеполовая, дерматологическая или иное нарушение, которое, по мнению следователя, могло повлиять на поведение следователя. исследование или всасывание, метаболизм или выведение исследуемого препарата. - Предыдущая история трансплантации костного мозга. - Заметное исходное удлинение интервала QT/QTc (например, повторная демонстрация QTc интервал > 480 миллисекунд [мсек]) (Общие критерии терминологии для нежелательных явлений [CTCAE] Степень 1) с использованием формулы коррекции интервала QT Фридериции. - Наличие в анамнезе дополнительных факторов риска TdP (например, сердечная недостаточность, гипокалиемия, семейное история синдрома удлиненного интервала QT). - Лечение цитотоксическими химиотерапевтическими агентами или экспериментальными агентами для лечения МДС в течение 8 недель исследуемого лечения. - Получение любой другой одновременной химиотерапии, лучевой терапии или иммунотерапии (в течение 8 недель до начала исследуемого лечения). - Использование сопутствующих препаратов, удлиняющих интервал QT/QTc во время исследуемого лечения. - Использование сопутствующих препаратов, которые являются сильными ингибиторами или индукторами CYP3A или CYP2B6. во время лечения
| Общий контакт | Контактная информация отображается только при наборе участников исследования. |
|---|
| Дата проверки |
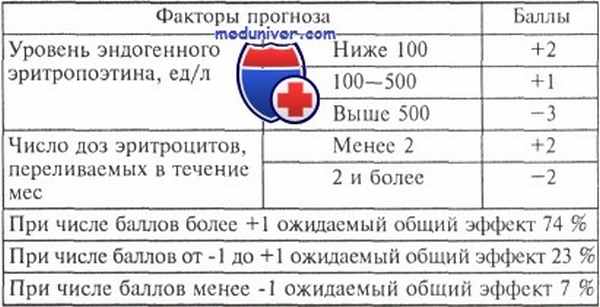
Метка: Увеличение дозы Описание: Стандартный дизайн 3+3 Экспериментальный: Фаза увеличения дозы Модель вмешательства: Одногрупповое задание Первичное назначение: Уход Маскировка: Нет (открытая этикетка) This information was retrieved directly from the website clinicaltrials.gov without any changes. If you have any requests to change, remove or update your study details, please contact [email protected] . As soon as a change is implemented on clinicaltrials.gov, this will be updated automatically on our website as well. Миелодиспластические синдромыВам поставили диагноз: Миелодиспластические синдромыНаверняка Вы задаётесь вопросом: что же теперь делать? Подобный диагноз всегда делит жизнь на «до» и «после». Все эмоциональные ресурсы пациента и его родных брошены на переживания и страх. Но именно в этот момент необходимо изменить вектор «за что» на вектор «что можно сделать». Очень часто пациенты чувствуют себя безгранично одинокими вначале пути. Но вы должны понимать - вы не одни. Предлагаем вашему вниманию краткий, но очень подробный обзор данного заболевания. Его подготовили высоко квалифицированные специалисты Отдела лекарственного лечения злокачественных новообразований МРНЦ имени А.Ф. Цыба и Отдела лекарственного лечения опухолей МНИОИ имени П.А. Герцена – филиалов ФГБУ «НМИЦ радиологии» Минздрава России под редакцией заведующих отделами, д.м.н. ФАЛАЛЕЕВОЙ Н.А. и д.м.н. ФЕДЕНКО А.А. Филиалы и отделения, где лечат миелодиспластические синдромыМНИОИ им. П.А. Герцена – филиал ФГБУ «НМИЦ радиологии» Минздрава России. Отдел лекарственного лечения опухолей тел: 8 (494) 150 11 22 МРНЦ им. А.Ф. Цыба – филиал ФГБУ «НМИЦ радиологии» Минздрава России. Отдел лекарственного лечения злокачественных новообразований тел: 8 (484) 399 – 31-30 Миелодиспластические синдромы(МДС) – разнородная группа заболеваний системы кроветворения, сопровождающаяся нарушением созревания кроветворных клеток с нарушениями их строения (дисплазией) и функции, а также повышенным риском развития острого лейкоза. МДС чаще всего сопровождаются снижением показателей общего анализа крови – цитопенией. Снижение уровня гемоглобина и числа эритроцитов обозначают термином анемия; числа лейкоцитов/нейтрофилов – лейкопенией/нейтропенией; уровня тромбоцитов – тробоцитопенией. Изредка может быть и увеличение числа лейкоцитов и/или тромбоцитов. Ежегодная заболеваемость МДС в среднем составляет 3-4 случая на 100000 населения и увеличивается с возрастом. Основной контингент больных МДС представлен пожилыми людьми (средний возраст – 70 лет). В отдельную группу относят МДС, развившиеся после химиотерапии и/или лучевой терапии предшествующих заболеваний (преимущественно онкологических). Они составляют 10-15% от ежегодно выявляемых случаев МДС и называются вторичными МДС. В связи с гетерогенностью заболевания возникает вопрос о выборе оптимальной терапии. Одним из основных факторов для решения этой задачи является определение прогностической группы (группы риска), к которой относится конкретный больной. Каждый прогностический признак оценивается в баллах. В соответствии с суммарным числом баллов больных объединяют в 4 группы: низкого (0 баллов), промежуточного-1 (0,5-1,0 балл), промежуточного-2 (1,5-2,0 балла) и высокого (2,5 балла и выше) риска. Первые 2 группы (низкого и промежуточного-1 риска) характеризуются принципиально благоприятным прогнозом, а 2 остальные (промежуточного-2 и высокого риска) — неблагоприятным прогнозом. Определение прогноза необходимо для выбора лечения. Существует несколько основных направлений в лечении МДС: Химиотерапия: на протяжении последних 20 лет для лечения больных МДС с увеличенным числом бластных клеток, то есть преимущественно в группе принципиально неблагоприятного прогноза, используются схемы терапии острого миелоидного лейкоза. Применение стандартной химиотерапии позволяет получить высокую частоту полных ремиссий – 50-70%. Однако продолжительность ремиссий относительно короткая (как правило, менее 1,5 лет), а лечение сопровождается высокой токсичностью. Трансплантация гемопоэтических стволовых клеток (трансплантация костного мозга): единственным методом лечения, позволяющим существенно увеличить продолжительность жизни больных МДС, является аллогенная трансплантация гемопоэтических стволовых клеток (аллоТГСК). Аллогенная ТГСК также позволяет получить наилучшие результаты по сравнению с другими методами лечения при вторичных МДС. Однако выполнение аллоТГСК не всегда возможно в связи с пожилым возрастом большинства больных и отсутствием идентичного родственного донора. Иммуносупрессивная терапия: использование иммуносупрессивных препаратов, таких как циклоспорин А, демонстрирует наибольшую активность при гипопластическом варианте МДС, при отсутствии увеличения числа бластных клеток в костном мозге, нормальном кариотипе (без отклонений в наборе хромосом), наличии лимфоидных узелков (очаговых скоплений лимфоидных клеток) в трепанобиоптате, наличии клона клеток, составляющих субстрат пароксизмальной ночной гемоглобинурии (ПНГ-клон), и у HLA-DR-15 – позитивных молодых больных. Терапия колониестимулирующими препаратами: эритропоэз-стимулирующие препараты – эритропоэтины (препараты, направленные на лечение анемии) фигурируют практически во всех рекомендациях по лечению МДС. Практически все специалисты сходятся во мнении о необходимости определения уровня эндогенного (собственного, вырабатываемого организмом) эритропоэтина до начала лечения. При уровне эндогенного эритропоэтина свыше 500 ед/л лечение эритропоэтином не показано, большая эффективность наблюдается при уровне эндогенного эритропоэтина в пределах 200 ед/л. Сопроводительная (симптоматическая) терапия : к сопроводительной терапии при МДС относят гемотрансфузии (переливания) эритроцитов и тромбоцитов, антимикробную терапию, в том числе в комбинации с Г-КСФ или ГМ-КСФ, использование комплексонов (хелаторов) железа. Программа лечения МДС основана на риск-адаптированном подходе в зависимости от групп прогноза, возраста, общего состояния больного. Иными словами, назначается индивидуальная терапия в зависимости от результатов полностью проведенного обследования. В некоторых случаях, когда проявления заболевания незначительны, может быть предложено только наблюдение у врача. Лечение некоторыми препаратами может проводиться как в условиях стационара, так и дома. Филиалы и отделения Центра, в которых лечат миелодиспластические синдромыФГБУ «НМИЦ радиологии» Минздрава России обладает всеми необходимыми технологиями лучевого, химиотерапевтического и хирургического лечения, включая расширенные и комбинированные операции. Все это позволяет выполнить необходимые этапы лечения в рамках одного Центра, что исключительно удобно для пациентов. Отдел лекарственного лечения злокачественных новообразований МРНЦ имени А.Ф. Цыба – филиал ФГБУ «НМИЦ радиологии» Минздрава России Заведующая отделом, д.м.н. ФАЛАЛЕЕВА Наталья Александровна 8 (484) 399 – 31-30, г. Обнинск, Калужской области Отдел лекарственного лечения опухолей МНИОИ имени П.А. Герцена –филиал ФГБУ «НМИЦ радиологии» Минздрава России Заведующий отделом, д.м.н. ФЕДЕНКО Александр Александрович Симптоматическая терапия миелодиспластического синдрома (МДС) - переливание эритроцитов, эритропоэтинСимптоматическая терапия миелодиспластического синдрома (МДС) - переливание эритроцитов, эритропоэтинБольшинство больных вне зависимости от варианта заболевания и принадлежности к той или иной прогностической группе нуждаются в симптоматической (сопроводительной) терапии. Этот вид лечения направлен на борьбу с осложнениями миелодиспластического синдрома, возникающими на протяжении заболевания. Проведение гемотрансфузий эритроцитов часто оказывается необходимым мероприятием при выраженной анемии (менее 80 г/л). При многократных переливаниях эритроцитов возникает опасность гемосидероза, что диктует необходимость применения комплексонов железа, например дефероксамина (десферала). Препарат следует назначать, по данным разных авторов, после 20—30 или после 50 гемотрансфузий эритроцитарной массы. Дефероксамин можно вводить как в дозе 2 г в день в виде 12-часовой подкожной инфузии (с помощью специального портативного дозатора), так и по 1 г в день в виде болюсного подкожного введения. Согласно рекомендациям NCCN, предпочтительным способом введения является подкожная инфузия, проводимая в ночное время. Помимо основного действия дефероксамина, при использовании его у больных миелодиспластическими синдромами описано уменьшение потребности в гемотрансфузиях, а в ряде случаев полное исчезновение необходимости переливания эритроцитов, увеличение числа тромбоцитов и нейтрофилов. Механизм уменьшения выраженности цитопении при терапии этим препаратом на настоящий момент остается неясным. При хронической кровопотере или дефиците железа, развивающегося на фоне лечения эритропоэтином, наоборот, могут возникать показания к назначению препаратов железа. Использование эритропоэтина (ЕРО) нередко позволяет нормализовать уровень гемоглобина или уменьшить частоту проведения гемотрансфузий. Применение ЕРО в онкологии, в том числе для лечения больных МДС, началось в конце 80-х годов XX в. К настоящему времени опубликовано большое количество работ, обобщающих результаты применения ЕРО более чем у 1000 больных МДС. G. Verhoef и соавт. приводят следующие данные: ответ на лечение ЕРО составляет 25 % (ни у одного больного не отмечено увеличения числа нейтрофилов, число тромбоцитов возросло в 1 % наблюдений, число бластных клеток повысилось в 0,7 % случаев). У большинства больных ответ на лечение был получен в течение первых 8 нед, однако описаны случаи достижения эффекта и через 18 нед. Зависимость эффективности лечения эритропоэтином больных миелодиспластическими синдромами от факторов прогнозаНаличие при миелодиспластическом синдроме двух- и трехростковой цитопении послужило основанием для применения комбинаций ростовых факторов. В основе совместного назначения различных цитокинов также лежит идея стимуляции дифференцировки клеток-предшественников на различных этапах их развития. В результате использования комбинации ЕРО с G- или GM-CSF, помимо увеличения числа нейтрофилов, был обнаружен синергизм препаратов при лечении анемии. Обобщая опыт использования ЕРО с G-CSF или GM-CSF, G. Verhoef и соавт. указывают на увеличение общего ответа со стороны красного ростка до 42—46 %, увеличение числа нейтрофилов в 79— 87% случаев, тромбоцитов — в 1—12%, бластных клеток в костном мозге — в 8 %. Представляет интерес тот факт, что при терапии EPO+G-CSF наиболее благоприятную группу составили больные РАКС (общий ответ получен в 52 %), которые реже больных с другими ФАБ-вариантами отвечают на терапию одним ЕРО. У части больных, не чувствительных к терапии ЕРО в монорежиме, после добавления G-CSF полностью нормализовался уровень гемоглобина. По данным R. Negrin и соавт., у половины больных, эффективно леченных EPO+G-CSF, после отмены G-CSF гематологический статус возвратился к исходному. На основании результатов двух многоцентровых исследований была разработана числовая система прогноза эффективности терапии EPO+G-CSF. По мнению G. Verhoef, достаточной дозой ЕРО для получения эффекта является 150 МЕ/кг 3 раза в неделю подкожно. В случае отсутствия ответа на лечение через 4—8 нед необходимо увеличить дозу ЕРО до 300 МЕ/кг 3 раза в неделю. В последнее время рекомендуется терапия более высокими дозами ЕРО. По мнению Е. Hellstrom-Lindberg, доза ЕРО должна составлять 50 000— 70 000 ME в неделю, разделенная на 3—5 введений. Терапия в этом режиме должна продолжаться не менее 6 нед. При отсутствии эффекта рекомендуется дополнительно назначить G-CSF. В том случае, если число нейтрофилов составляет менее 1,5•10 9 /л, G-CSF применяется в дозе, поддерживающей их нормальный уровень. При числе нейтрофилов более 1,5•109/л препарат используют в дозе, позволяющей увеличить их уровень в 2 раза. Как правило, эффект от комбинированного лечения наступает в течение 3 мес. Недавно были опубликованы предварительные результаты рандомизированного исследования терапии ЕРО в монорежиме (по 10 000 ME 3 раза в неделю подкожно) и в комбинации с G-CSF (этот же режим применения EPO+G-CSF по 5 мкг/кг 2 раза в неделю подкожно) у больных МДС с благоприятным прогнозом по шкале IPSS. Вероятность ответа больных миелодиспластическими синдромами на лечение EPO+G-CSF
Примечание. Общий эффект объединяет полный ответ на лечение (повышение уровня гемоглобина до 115,0 г/л) и частичный (повышение уровня гемоглобина на 15,0 г/л или исчезновение показаний к гемотрансфузиям эритроцитов). Через 2 мес лечения эффективность ЕРО в монорежиме и в комбинации с G-CSF составила 41,6 % и 75 % соответственно. К 6 мес лечения эффект сохранялся у 33 % больных первой группы и у 58 % — второй. Число больных, у которых произошла лейкемическая трансформация, было одинаковым в обеих группах — 16 %. Учитывая накопленный опыт терапии ЕРО, NCCN предложило алгоритм лечения анемии. 3) При исходном уровне эндогенного ЕРО > 500 ед/л лечение эритропоэтином не показано. В настоящее время накапливается опыт применения гипергликозилированного человеческого рекомбинантного ЕРО (дарбепоэтина) у больных миелодиспластическими синдромами. Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021 Миелодиспластический синдром (МДС)Миелодиспластический синдром (МДС) представляет собой группу заболеваний, характеризующихся цитопенией в периферической крови, дисплазией гемопоэтических клеток-предшественников, гиперклеточностью или гипоклеточностью костного мозга и высоким риском развития острого миелолейкоза Острый миелолейкоз (ОМЛ) При остром миелолейкозе (ОМЛ) злокачественная трансформация и неконтролируемая пролиферация аномально дифференцированных, долго живущих клеток-предшественниц миелоидного ряда вызывает появление. Прочитайте дополнительные сведенияЕжегодное число людей в Соединенных Штатах с диагнозом миелодиспластический синдром (МДС) неизвестно. Согласно некоторым оценкам, это число составляет около 10 000, в то время как по другим оценкам оно намного выше. МДС чаще всего диагностируется у пациентов в возрасте 70 лет. Патофизиология МДСМиелодиспластические синдромы представляют собой группу заболеваний клональных гемопоэтических стволовых клеток, объединенных наличием различных мутаций гематопоэтических стволовых клеток, чаще всего в генах, участвующих в сплайсинге РНК. Миелодиспластические синдромы характеризуются неэффективным и диспластическим гемопоэзом и включают в себя следующее: Рефрактерная анемия: анемия с ретикулоцитопенией; нормальный или гиперклеточный костный мозг с эритроидной гиперплазией и дизэритропоэзом; содержание бластных клеток составляет ≤ 5% ядросодержащих клеток костного мозга Рефрактерная анемия с кольцевыми сидеробластами: то же, что и рефрактерная анемия с ретикулоцитопенией, за исключением того, что кольцевые сидеробласты составляют > 15% ядросодержащих клеток костного мозга Рефрактерная цитопения с мультилинейной дисплазией: цитопения не определяется только эритроцитами;имеет место выраженная дисплазия предшественников лейкоцитов и мегакариоцитов Рефрактерная цитопения с мультилинейной дисплазией и кольцевыми сидеробластами с наличием кольцевых сидеробластов, которые составляют > 15% ядросодержащих клеток костного мозга Рефракторная анемия с избытком бластов (РАИБ) (RAEB en.): цитопения ≥ 2 клеточных линий с морфологическими аномалиями гематопоэтических клеток; гиперцеллюлярный костный мозг с дизэритропоэзом и дисгранулопоэзом; разрушает от 5 до 9% (RAEB-I) или от 10 до 19% (RAEB-II) ядросодержащих клеток костного мозга. Миелодиспластический синдром неклассифицированный: МДС, который не попадает ни в одну из определенных категорий МДС с изолированной делецией 5q: обычно тяжелая анемия и тромбоцитоз с делецией длинного плеча пятой хромосомы. Хронический миеломоноцитарный лейкоз (ХММЛ) и ювенильный миеломоноцитарный лейкоз (ЮММЛ): смешанные миелодиспластические/миелопролиферативные новообразования; абсолютный моноцитоз (> 1000/мкл [> 1/л]) крови; значительное увеличение количества предшественников моноцитов в костном мозге Хронический нейтрофильный лейкоз: характеризуется нейтрофилией, гибридным геном BCR-ABL1 и отсутствием филадельфийской хромосомы. Этиология миелодиспластического синдрома неизвестна. Риск повышается с возрастом из-за приобретенных соматических мутаций, которые могут способствовть клональной экспансии и доминированию определенных гемопоэтических стволовых клеток, и, возможно, посредством воздействия внешних токсинов, таких как бензин, ионизирующие излучение и химиотерапевтические препараты (особенно продолжительные или интенсивные курсы лечения, а также с использованием алкилирующих агентов, гидроксимочевины или ингибиторов топоизомеразы). Часто присутствуют хромосомные аномалии (например, делеции, дупликации, структурные аномалии). Костный мозг может быть гиперклеточным или гипоклеточным Неэффективный гемопоэз приводит к анемии (встречается наиболее часто), нейтропении, тромбоцитопении, или к комбинации этих патологий, вплоть до аплазии костного мозга. У пациентов со значительной рефрактерной или хронической анемией в конечном итоге развивается перегрузка железом ввиду переливания крови и/или повышенной абсорбции железа с кишечника. Нарушение клеточной продукции также сопровождается изменениями морфологии клеток в костном мозге и крови. Иногда развивается экстрамедуллярный гемопоэз, приводящий к гепатомегалии и спленомегалии. Во время МДС может развиваться миелофиброз Первичный Миелофиброз Первичный миелофиброз (ПМФ) – это хроническое миелопролиферативное новообразование, которое характеризуется фиброзом костного мозга, спленомегалией и анемией с наличием ядросодержащих и каплевидных. Прочитайте дополнительные сведения . Классификация основана на данных общего анализа крови и исследований костного мозга, а также учитывается кариотип и мутация. Клон МДС имеет тенденцию к трансформации в острый миелолейкоз Острый миелолейкоз (ОМЛ) При остром миелолейкозе (ОМЛ) злокачественная трансформация и неконтролируемая пролиферация аномально дифференцированных, долго живущих клеток-предшественниц миелоидного ряда вызывает появление. Прочитайте дополнительные сведенияСимптомы и признаки МДССимптомы миелодиспластического синдрома зависят от наиболее пораженной клеточной линии и могут включать бледность, слабость и утомляемость (анемия), лихорадку и инфекции (нейтропения), повышенную склонность к кровоизлияниям, петехиям и кровоточивости из слизистых оболочек (тромбоцитопения). Спленомегалия и гепатомегалия не редкость. Миелодиспластический синдромМиелодиспластический синдром (МДС) – группа гематологических заболеваний, обусловленных нарушением работы костного мозга по воспроизведению одного или более типов клеток крови: тромбоцитов, лейкоцитов, эритроцитов. У людей, страдающих МДС, костный мозг, компенсируя естественный процесс уничтожения клеток крови селезенкой, не в состоянии воспроизвести их в нужном количестве. Это приводит к увеличению риска инфекций, кровоточивости и анемии, которая также проявляется усталостью, одышкой или сердечной недостаточностью. Развитие МДС может носить как спонтанный характер (без видимых причин), так и быть обусловленным использованием химиопрепаратов, облучения. Последний вариант МДС часто называют «вторичным», и хотя встречается он гораздо реже, хуже поддается лечению. Подавляющее большинство случае «первичного» МДС развивается у людей старше 60 лет, в детском возрасте болезнь встречается редко. Клиническая картина МДС В подавляющем большинстве пациенты обращаются за помощью с жалобами на усталость, утомляемость, одышку при физической нагрузке, головокружения – симптомами, связанными с развитием анемии. Остальным же пациентам диагноз устанавливается случайно, при лабораторном тестировании анализов крови, сделанных по другим причинам. Реже диагноз устанавливается при лечении инфекции, геморрагического синдрома, тромбоза. Такие признаки, как потеря веса, немотивированная лихорадка, болевой синдром, также могут быть манифестацией МДС. Диагностика МДС базируется, прежде всего, на лабораторных данных, которые включают:
Обязательные диагностические мероприятия при МДС В обязательный перечень диагностических мероприятий входят:
Хромосомные нарушения подтверждают присутствие патологического клона и являются определяющими при решении вопроса о наличии МДС или реактивных изменений. Классификация МДС основывается на количестве и типе бластных клеток, а также наличии хромосомных изменений, при этом тип МДС у пациента по мере развития заболевания может изменяться в сторону прогрессирования, вплоть до развития острого миелобластного лейкоза у 10% пациентов. Это система классификации используется ВОЗ. Возможно, самая полезная система клинической классификации для МДС – Международная Прогностическая Система (IPSS). Эта модель была разработана для оценки таких переменных категорий как возраст, тип бластных клеток, генетические изменения. На основании этих критериев было выявлено 4 группы риска – низкий, промежуточный 1, промежуточный 2 и высокого риска. Рекомендации по лечению основываются именно на отношении пациента к какой-либо из групп риска. Так пациент с низким уровнем риска может жить много лет прежде, чем потребуется лечение МДС, в то время как человек с промежуточным или высоким риском обычно нуждается в немедленном начале лечения. Всемирная организация здравоохранения (ВОЗ), основываясь на уровнях доказательности, издала предложения о новой классификации МДС.
В настоящее время нет никакого иного способа радикального лечения МДС кроме пересадки костного мозга, хотя существует множество схем для контроля симптомов, осложнений и улучшения качества жизни. Рекомендации NCCN предлагают, чтобы выбор лечения был основан на возрасте пациента, оценке возможности пациента выполнять нормальные ежедневные задачи и группе риска.
Лечение низкой интенсивности Поддерживающая терапия является важной частью лечения, и учитывает, как правило, пожилой возраст пациентов, она включает симптоматическую терапию, направленную на поддержание уровня лейкоцитов, тромбоцитов, эритроцитов. Эта терапия призвана улучшить качество жизни и продлить её продолжительность.
Комбинированная химиотерапия, используя G-CSF наряду с EPO, может быть более эффективной, чем использование одного только EPO, особенно у людей из низкой группы риска при пониженном фоновом уровне EPO в сыворотке. Иммунодепрессивные препараты могут быть эффективны у пациентов с гипопластическим типом кроветворения. Некоторые из этих пациентов, особенно молодые с ранней стадией болезни и гипоплазией, отвечают на иммунодепрессивные методы лечения, которые противостоят иммунной атаке на костный мозг. Использование иммунодепрессивной терапии может позволить 50-60% пациентам с HLA DR2 типом ткани прекратить заместительную терапию. Схемы иммунодепрессивных методов лечения включают антитимоцитарный глобулин (ATG) и циклоспорин. ATG обычно используется в качестве внутривенной инфузии один раз в день в течение 4 дней, в то время как циклоспорин обычно назначает перорально (приём таблеток) на длительное время, до развития тяжелых осложнений или прогрессирования МДС на фоне лечения. Наиболее частыми осложнениями терапии ATG можно считать сывороточную болезнь, купируемую назначение стероидных гормонов. Производные талидомида – препарата, стимулирующего иммунную систему и его аналоги (Revlimid ® , lenalidomide), – успешно используются в лечении других гемобластозов (лимфомы, множественная миелома). Lenalidomide особенно эффективен у пациентов с анемией из низкой или промежуточной 1 групп МДС с повреждением 5 хромосомы (синдром 5q минус). Низкие дозы цитостатиков в монорежиме могут быть рекомендованы для людей с промежуточным или высоким риском, которые не являются кандидатами для высокодозной терапии в силу различных причин.
Терапия МДС высокой интенсивности Пациенты с промежуточным или высоким риском при МДС подлежат терапии режимами химиотерапии аналогично используемым для лечения острого миелобластного лейкоза ОМЛ. Однако это лечение рекомендуется относительно молодым людям (моложе 60 лет), с хорошим жизненным статусом и при отсутствии HLA-идентичного донора. Этот тип лечения лучше не применять у лиц старше 60 лет, а также при низком жизненном статусе или большом числе цитогенетических нарушений, так как сопряжен с тяжелыми осложнениями. У некоторых пациентов поддерживающая терапия может обеспечить тот же результат, что и химиотерапия, но с более низким риском осложнений или токсичности. Некоторые пациенты добиваются большего успеха при проведении только симптоматической терапии осложнений МДС (анемии, инфекции, кровоточивости), не пытаясь излечить саму болезнь. Как ранее упоминалось, трансплантация стволовых клеток является единственным видом лечения, который может привести к длительной ремиссии. Однако осложнения терапии могут превалировать над возможным эффектом. В прошлом пациентов старше 50 лет не рассматривали как кандидатов для такого лечения. Достижения последних пятнадцати лет позволили отодвинуть возрастную планку до 60 лет и старше. Однако приблизительно 75% пациентов с МДС на момент установки диагноза уже старше 60 лет, таким образом, обычная трансплантация может быть предложена только малой части пациентов. Трансплантация рекомендуется для людей с промежуточным 1, промежуточными 2 и высоким риском моложе 60 лет и наличием идентичного донора, но не для пациентов с низкой группой риска. Хотя есть существенный шанс на получение ремиссии у пациентов из группы риска (60%), весьма высоки (более 40%) связанные с пересадкой смертельные случаи и частота рецидивирования в течение 5 лет. Возможно использование неродственных доноров, но в этой ситуации возраст пациента является важным фактором в успехе лечения. Использование режимов пониженной интенсивности при трансплантации расширяет категории пациентов, которым можно провести это лечение, но долгосрочные результаты еще нуждаются в оценке. Пока создается впечатления о повышенной частоте рецидивов по сравнению со стандартной подготовкой к трансплантации. У пациентов с МДС средняя продолжительность жизни зависит от категории риска и возраста. Есть значительные вариации течения болезни от пациента к пациенту, особенно в группе низкого риска. Руководитель отделения гематологии Читайте также:
|
|---|