Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Ген FGFR2 (fibroblast growth factor receptor 2) расположен на хромосоме 10 в регионе 10q26.13. Содержит 20 экзонов. Известно 26 мутаций, в основном располагающихся в экзонах 7 и 9 гена, приводящих к развитию заболевания.
Мутации в данном гене приводят также к развитию: синдрома Аперта, синдрому морщинистой кожи Беаре-Стевенсона, синдрома дисплазии Bent bone, краниофациально-скелетно-дерматологической дисплазии, неспецифическому краниосиностозу, синдрому Джексона-Вейсса, синдрому LADD, синдрому Антли-Бикслера, синдрому Крузона, синдрому Сетре-Чотзена, скафоцефалии и аномалии Аксенфельда-Ригера, синдрому скафоцефалии, западения верхней челюсти и умственной отсталости, соматическая мутация приводит к раку желудка.
Мутации в данном гене приводят также к развитию синдрома Джексона-Вейсса, гипогонадотропному гипогонадизму тип 2 с/без аносмии, остеоглофонической дисплазии, тригоноцефалии тип 1.
Заболевания, основными клиническими проявлениями которого являются краниосиностоз, широкие большие пальцы кистей и стоп, частичная синдактилия мягких тканей кистей рук.
Акроцефалия - основной и постоянный признак синдрома, но краниостеноз при этом отмечается не всегда. Возможен синостоз коронарного, сагиттального и ламбдовидного швов. В отдельных случаях наблюдается асимметрия черепа.
Черепно-лицевые дисморфии сходны с таковыми при синдроме Аперта: уплощенная спинка носа, гипоплазия верхнечелюстных костей, гипертелоризм, антимонголоидный разрез глазных щелей, высокое арковидное небо, сверхкомплектные зубы. Вместе с тем каждый из названных признаков встречается реже, чем при синдроме Аперта, и выраженность их меньше. Расщелины неба встречаются в 14% случаев. Экзофтальм и косоглазие выявляются примерно с той же частотой.
В части случаев при синдроме Пфайффера отмечают необычную патологию черепа - в форме трилистника или листа клевера.
Поражение конечностей обычно сводится к резкому расширению первых пальцев кистей и стоп, частичной синдактилии кистей (73%) и стоп (82%). Синдактилия при синдроме Пфайффера никогда не бывает тотальной, как при синдроме Аперта.
Характерен нормальный интеллект, но дальнейшее его развитие во многом определяется адекватным по срокам оперативным вмешательством. В редких случаях у больных обнаруживается гидроцефалия и мальформация Арнольда Киари, микрополигирия, аплазия мозолистого тела, недоразвитие островков Гейла. Другие аномалии представлены: патологией костей таза, coxa valga, пилоростенозом, пупочной грыжей, патологией лор-органов, тугоухостью, частичной атрофией зрительных нервов.
I тип синдрома Пфайфера характеризуется классическим проявлением краниосиностоза, широкими большими пальцами, синдактилией и нормальным интеллектом. Эта форма совместима с жизнью.
При II типе наблюдают череп в виде листка клевера, экзоорбитизм, широкие большие пальцы рук, анкилоз локтевого сустава, сниженный интеллект и серьезные нарушения центральной нервной системы (ЦНС). В результате обзора литературы по всем типам синдрома Пфайфера установлено, что большинство больных II типа умирают вскоре после рождения от дыхательной недостаточности, аномалий головного мозга, недоношенности или послеоперационных осложнений.
При III типе наблюдается краниосиностоз, выраженный экзоорбитизм, без черепа в форме трилистника, локтевой анкилоз и различные серьезные нарушения ЦНС. Прогноз негативный, т.к. заболевание приводит к ранней смерти.
Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
Существует три типа аномалии Арнольда-Киари (Arnold-Chiari). При типе I вниз смещаются только губа мозжечка с миндалинами, но четвертый желудочек остается локализован в задней черепной ямке. В большинстве наблюдений это случайно обнаруживается при проведении пациентам компьютерной томографии. Тип II обычно диагностируется пренатально и является пороком развития, который характеризуется смещением миндалин и части мозжечка, четвертого желудочка, варолиева моста и продолговатого мозга через большое затылочное отверстие вниз и внутрь спинномозгового канала. Эти изменения обычно сочетаются с наличием гидроцефалии и менингомиелоцеле. Тип III является наиболее тяжелой формой аномалии с большим объемом протрузии содержимого задней черепной ямки в спинномозговой канал в сочетании с менингомиелоцеле и гидроцефалией.
Для описания деформации мозжечка при эхографическом исследовании предложено использовать термин симптом «банана».
Обычно сочетается с расщелиной позвоночника (spina bifida), в противном случае встречается редко.
Аутосомно-рецессивный тип наследования некоторых форм.
Смещение мозжечка деформирует его латеральные доли (которые утрачивают округлую форму) и инцизура мозжечка начинает выглядеть более сглаженной (что обусловливает появление «симптома банана»). Диагноз может быть установлен начиная с первого триместра беременности.
Дифференциальный диагноз. Необструктив-ные формы гидроцефалии.
Сочетанные аномалии. Гидроцефалия вследствие обструкции отверстия Маженди (Magendie), си-рингомиелия и дистематомелия.
Прогноз. Неблагоприятный вследствие наличия аномалий ЦНС.
Акушерская тактика. До наступления периода жизнеспособности плода может быть предложено прерывание беременности. При выявлении расщелин позвоночника (spina bifida) в нескольких прена-тальных центрах в США предлагалось экспериментальное внутриутробное хирургическое вмешательство, осуществляемое в сроки от 21 до 30 нед (в США звонить по номеру: 800-RX-FETUS, а также в Vanderbilt University). Если принимается решение о пролонгировании беременности, стандартная тактика пренатального ведения пациентки не изменяется. Важное значение для генетического консультирования семейной пары имеет подтверждение диагноза после рождения.
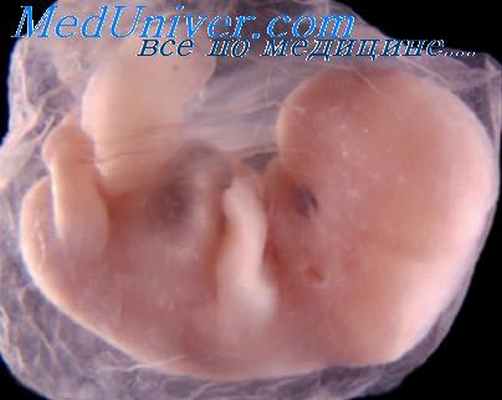
Множественный врожденный артрогрипоз плода
Данное заболевание представляет собой ряд гетерогенных состояний, основными признаками которых являются ограничение объема движений и анкилозы суставов различной степени выраженности.
Синонимы. Врожденные контрактуры, секвенция акинезии плода, синдром Пена-Шокейра (Репа-Shokeir).
Распространенность. Примерно 1-3 случая на 10 000 родов.
Этиология. Возможно, аутосомно-доминантный тип наследования.
Патогенез. Артрогрипоз возникает вследствие снижения возможности у плода проявлять внутриутробную двигательную активность в результате наличия поражений неврологического, мышечного, соединительнотканного или инфекционного генеза.
Несмотря на то что аномалии бывают достаточно очевидны, когда выявляются при эхографическом обследовании и особенно при осмотре после рождения ребенка, пренатальная диагностика может быть затруднена в случаях уменьшения объема околоплодных вод, то есть в ситуациях, при которых патологическое положение конечностей может быть расценено как следствие маловодия. При некоторых формах артрогрипоза наблюдается многоводие, и тогда аномальное положение конечностей (сгибание коленного сустава под углом, открытым латерально или кпереди, а также косолапость и косорукость) позволяет легко устанвить диагноз. Многоводие часто является следствием уменьшения заглатывания плодом околоплодных вод, что также может быть частью патогенеза артрогрипоза (что связано с мышечными или неврологическими нарушениями). В первом триместре у таких плодов могут выявляться увеличение толщины воротникового пространства и характерное снижение двигательной активности.
Генетические нарушения. Некоторые аномалии связаны с дефектами, локализованными в хромосомах 5q35, 9p21-q21 и 11р15.5.
Дифференциальный диагноз. Часть признаков при артрогрипозе может быть аналогичной при три-сомии 18, агенезии почек и миотонической дистрофии.
Сочетанные аномалии. Вследствие гетерогенности проявлений заболевания описывалось множество сочетанных аномалий, включая сколиоз, аномалии ЦНС и даже судороги.
Прогноз. Прогноз будет зависеть от наличия сочетанных аномалий (дыхательных нарушений, сколиоза и других).
Акушерская тактика. До наступления периода жизнеспособности плода может быть предложено прерывание беременности. Если принимается решение о ее пролонгировании, стандартная тактика пренатального ведения пациентки не изменяется. Большое значение для генетического консультирования семейной пары имеет подтверждение диагноза после рождения.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Множественный врождённый артрогрипоз
Множественный врожденный артрогрипоз связан с рядом состояний, включающих врожденные ограничения подвижности суставов. Интеллект, как правило, нормальный, за исключением случаев, когда артрогрипоз вызван заболеванием или синдромом, также влияющим на интеллект. Диагноз стаавится на основе клинических данных Лечение включает манипуляции с суставом и формирование, а иногда и хирургическое вмешательство.
Артрогрипоз – это не конкретный диагноз, а скорее клинический вывод о наличии врожденных контрактур, которые могут наблюдаться при более чем 300 различных нарушениях. Распространенность в различных исследованиях варьирует примерно между 1/3000 и 1/12 000 случаев у живорожденных детей. Перинатальная смертность при некоторых из скрытых состояний составляет 32%, поэтому установления конкретного диагноза является важным для прогноза и генетического консультирования.
Существует два основных типа множественного врожденного артрогрипоза (МВА):
Амиоплазия (классический артрогрипоз): в конечностях возникают множественные симметричные контрактуры. Пораженные мышцы гипопластичны с наличием фиброза и жировой дистрофии. Обычно уровень интеллекта нормальный. Около 10% пациентов имеют аномалии брюшной полости (например, гастрошизис Гастрошизис Гастрошизис – выпячивание внутренних органов брюшной полости через дефект брюшной стенки; обычно локализируется справа от места отхождения пуповины. (См. также Обзор врожденных аномалий желудочно-кишечного. Прочитайте дополнительные сведенияДистальный артрогрипоз: Вовлечены верхние и нижние конечности, но крупные суставы обычно не затронуты. Дистальные артрогрипозы представляют собой гетерогенную группу расстройств, многие из которых связаны с определенным генетическим дефектом, выявленным среди ряда генов, которые кодируют компоненты сократительного аппарата. Дистальные артрогрипозы передаются как аутосомно-доминантные заболевания, но известны также Х-сцепленные мутации.
Этиология
Любое состояние, ухудшающее внутриутробные движения на > 3 недели, может привести к МВА. Причинами могут являться:
Заболевания матери (например, рассеянный склероз Рассеянный склероз (РС) Рассеянный склероз (РС) характеризуется появлением в головном и спинном мозге диссеминированных очагов демиелинизации. Характерные симптомы включают зрительные и глазодвигательные нарушения. Прочитайте дополнительные сведения Более 35 специфических генетических нарушений (например, спинальная мышечная атрофия типа I, трисомия по 18 хромосоме Трисомия по 18 хромосоме Трисомия по 18 хромосоме вызывается дополнительной 18 хромосомой и, как правило, связана с умственной отсталостью, малым весом при рождении, а также различными врожденными аномалиями развития. Прочитайте дополнительные сведенияКлинические проявления
Деформации заметны при рождении. МВА не прогрессирует; однако, состояния, которые его вызывают (например, мышечная дистрофия), могут быть прогрессирующими. Пораженные суставы сжимаются при сгибании или разгибании. При классических проявлениях МВА плечи покатые, приведены и ротированы внутрь; локти вытянуты; запястья и пальцы согнуты. Бедра могут быть дислоцированы и, как правило, слегка согнуты. Колени удлинены; ноги часто находятся в эквиноварусной позиции. Мышцы ног, как правило, гипопластичные, конечности цилиндрические и не имеют четких контуров. Иногда возникает перепончатость мягких тканей вдоль вентральной стороны согнутых суставов. Позвоночник может быть сколиозным. Несмотря на тонкость длинных костей, на рентгенограмме скелет выглядит нормальным. Физические недостатки могут быть тяжелыми. Как уже отмечалось, некоторые дети могут иметь первичную дисфункцию центральной нервной системы, но интеллект обычно не затронут.
Интубация трахеи во время операции может быть сложной, потому что у детей недоразвита челюсть и височно-нижнечелюстное сочленение. Другие аномалии, которые редко сопровождают артрогрипоз, включают микроцефалию, волчью пасть, крипторхизм, а также аномалии сердца и мочевыводящих путей; наличие этих аномалий вызывает подозрения на скрытый хромосомный дефект или генетический синдром.
Диагностика
Обследование для выяснения причины
Если у новорожденного присутствует несколько контрактур, задача первоначальной оценки – определить является ли состояние амиоплазией, дистальным артрогрипозом или другим синдромом, при котором несколько контрактур связаны с другими врожденными аномалиями и/или метаболическими нарушениями. Если это возможно, клинический генетик должен координировать оценку и лечение; как правило, в данном процессе задействованы практикующие врачи многих специальностей. Синдромная форма МВА подозревается, когда присутствуют задержки в развитии и/или другие врожденные аномалии; таких пациентов необходимо обследовать на наличие расстройств центральной нервной системы, также нужно проводить мониторинг нарастания неврологических симптомов.
Оценка также должна включать тщательную проверку ассоциированных физических, хромосомных и генетических аномалий. Конкретные расстройства следует искать среди следующих: синдром Фримена-Шелдона, синдром Холта-Oрама, синдром Ларсена, синдром Миллера, синдром множественных птеригиумов и синдром Ди Джорджи (синдром делеции 22q11). Исследование обычно начинают с хромосомного микроматричного анализа с последующими специфическими генетическими тестами, которые проводят по отдельности или в стандартной панели во многих генетических лабораториях ( 1 Справочные материалы по диагностике Множественный врожденный артрогрипоз связан с рядом состояний, включающих врожденные ограничения подвижности суставов. Интеллект, как правило, нормальный, за исключением случаев, когда артрогрипоз. Прочитайте дополнительные сведения Полноэкзомное секвенирование должно применяться когда другие исследования не дают окончательного диагноза, особенно в семейных случаях ( 2 Справочные материалы по диагностике Множественный врожденный артрогрипоз связан с рядом состояний, включающих врожденные ограничения подвижности суставов. Интеллект, как правило, нормальный, за исключением случаев, когда артрогрипоз. Прочитайте дополнительные сведенияСправочные материалы по диагностике
1. Todd EJ, Yau KS, Ong R, et al: Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis 10:148, 2015. doi: 10.1186/s13023-015-0364-0.
2. Hunter JM, Ahearn ME, Balak CD, et al: Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol Genet Genomic Med 3(4):283–301, 2015. doi: 10.1002/mgg3.142.
Синдром Арнольда-Киари. Множественный врожденный артрогрипоз плода
АНОМАЛИЯ АРНОЛЬДА-КИАРИ ВО ВРЕМЯ БЕРЕМЕННОСТИ: АНЕСТЕЗИОЛОГИЧЕСКИЕ АСПЕКТЫ
Е.М.Шифман, А.В.Куликов
ОСНОВНЫЕ ПОНЯТИЯ.
Определение
■ Аномалия Арнольда-Киари – врожденная патология, характеризующаяся опущением миндаликов мозжечка в большое затылочное отверстие.
■ Выделяют 4 степени аномалии Арнольда-Киари; чаще встречаются I и II степень.
I степень. Характеризуется удлинением и каудальным смещением миндаликов мозжечка ниже большого затылочного отверстия.
II степень. Каудальное смещение миндаликов мозжечка нижнего червя, четвертого желудочка и продолговатого мозга. Дисплазия остальных отделов мозжечка, спинной мозг часто удлиненный и извилистый.
III степень. Каудальное смещение мозжечка и большого мозга в высокое шейное миеломенингоцеле.
IV степень. Гипоплазия мозжечка с грыжеобразованием.
Эпидемиология
♦ Обычно диагностируется у лиц молодого возраста.
♦ Обычно проявляется в детстве и юности.
Патофизиология
♦
Редко развивается блок циркуляции СМЖ, с формированием несообщающейся гидроцефалии.
♦ Аномалии тела шейных позвонков (обычно первого шейного) имеются приблизительно 5% случаев, очень редко эти нарушения сочетаются с миеломенингцеле.
♦ Обычно имеются spina bifida, гидроцефалия и сирингомиелия.
♦ Лечение заключается в коррекции миеломенингоцеле и наложении шунта для купирования гидроцефалии.
Клинические проявления
♦ Характерные симптомы — головная боль, провоцируемая приемом Вальсавы, боли в области шеи и верхних конечностей, мозжечковая симптоматика (включая атаксию, головокружение).
♦ Возможны слабость в конечностях и расстройства чувствительности (онемение).
■ Как правило, если миндалики опущены в большое затылочное отверстие более чем на 5 мм, имеются клинические проявления поражения нервной системы.
♦ Нистагм, стридор, апноэ, снижение глоточного рефлекса, слабость в верхних конечностях.
Влияние беременности на течение заболевания
■ Прием Вальсавы в первом периоде родов может привести к значительномуухудшению неврологической симптоматики, вплоть до утраты сознания.
Влияние заболевания на течение беременности и состояние плода
■ Не отмечено отрицательного влияния на течение беременности и состояние плода.
ИССЛЕДОВАНИЯ
■ Диагноз ставится при помощи нейровизуализационной методики (предпочтительно МРТ).
ЛЕЧЕНИЕ
■ Лечение чаще консервативное, может потребоваться хирургическая декомпрессия или
наложение
шунта при наличии выраженной гидроцефалии.
Хирургическое лечение
■ При отсутствии клинических проявлений хирургическое лечение не показано.
■ В случае прогрессирования тяжелой симптоматики – субокципитальная краниотомия с пластикой дефекта твердой мозговой оболочкой.
♦ В сочетании с миеломенингоцеле (II степень) может быть показано хирургическое лечение.
♦ В
сочетании с гидроцефалией (II степень) может потребоваться наложение шунта.
Особенности анестезии
■ Применяется и общая анестезия и регионарные методы обезболивания – без осложнений, как с проведенной хирургической декомпрессией, так и без нее.
■ Если пациентке была проведена хирургическая декомпрессия риск грыжеобразования (вклинения) низкий. Возможна регионарная анестезия (спинальная или эпидуральная) или быстрая последовательная индукция при предстоящей общей анестезии.
■ Если
внутричерепное давление высокое, а хирургическая декомпрессия не проводилась, то пункция твердой мозговой оболочки может привести к дальнейшему опущению миндаликов мозжечка в большое затылочное отверстие и ухудшению неврологического статуса.
♦ Есть данные о впервые диагностированной аномалии Арнольда-Киари после пункции твердой мозговой оболочки (возникновение неврологических симптомов: головной боли, нарушений зрения и слуха).
♦ В то же время ,есть описания случаев и описания серии случаев о неосложненных спинальных и эпидуральных анестезиях у пациенток с неврологической симптоматикой при аномалии Арнольда-Киари.
ПРЕДУПРЕЖДЕНИЯ И РЕКОМЕНДАЦИИ
■ Аномалия Арнольда-Киари – врожденная патология, характеризующаяся опущением миндаликов мозжечка в большое затылочное отверстие.
■ Выделяется четыре степени аномалии Арнольда-Киари; чаще встречается I и II степень.
■ Симптомы (по снижению частоты): боли в шейно-затылочной области, нарушения зрения и отоневрологические симптомы, атаксия, головокружение, слабость и онемение в конечностях.
■ Диагноз ставится при использовании нейровизуализационных методов – предпочтительна МРТ.
■ Как регионарная, так и общая анестезия может успешно применяться у пациенток с аномалией Арнольда-Киари как после проведения декомпрессии, так и без нее.
■ После выполнения хирургической декомпрессии риск грыжеобразования (вклинения) низкий.
■ При высоком внутричерепном давлении после пункции твердой мозговой оболочки возможно дальнейшее опущение миндаликов мозжечка в большое затылочное отверстие и усугубление неврологической симптоматики у пациенток, которым хирургическая декомпрессия не выполнялась.
ОБЯЗАТЕЛЬНЫЕ ДЕЙСТВИЯ
♦ Оценить возможность приема Вальсавы во время родоразрешения – при противопоказаниях обсудите с акушером способ вагинального родоразрешения с исключением потужного периода или кесарева сечения.
♦ Продумать в каждом конкретном случае приведет ли пункция твердой мозговой оболочки к прогрессированию процесса, спросить нейрохирурга – выполнил бы он пункцию у такой пациентки.
■ Выполнить МРТ для определения наличия и локализации сирингомиелитических изменений до выполнения
nрегионарной анестезии.
Редкие болезни
Италия – красивейшая страна, которой удалось создать одну из самых лучших в мире систем здравоохранения. По мнению известного международного агентства BLOOMBERG Италия уверенно входит в тройку мировых лидеров в медицине, уступая только Гонконгу и Сингапуру, и оставляя далеко за собой систему здравоохранения Германии.
Список редких заболеваний, которые лечатся в Италии
1. Адренолейкодистрофия
2. Врожденные аномалии метаболизма липопротеидов
3. Другие множественные врожденные аномалии, приводящие к умственной отсталости
4. Первичный амилоидоз и подобное
5. Наследственные анемии
6. Множественный врожденный артрогрипоз
7. Атрофия зрительного нерва
8. Атрофия мышц спины
9. Хронические воспалительные демиелинизирующие понейропатии
10.Первичный склерозирующий холангит
11.Врожденные хондродистрофии
12.Хорея Гентингтона
13. Смешанная криоглобулинемия
14. Дефицит АКТГ
15. Дерматомиозит
16. Нефрогенный несахарный диабет
17. Наследственные дефекты коагуляции
18. Наследственные дистрофии хориоидеи
19. Миотоническая дистрофия
20. Мышечные дистрофии
21. Наследственные дистрофии сетчатки
22. Накопление липидных нарушений
23. Нарушения метаболизма и транспорта углеводов, за исключением Сахарного диабета
24. Врожденная слепота
25. Мохроматоз
26. Пароксизмальная ночная Гемоглобинурия
27. Ревматический эндокардит
28. Истинный гермафродитизм
29. Эозинофильный фасцит
30. Забрюшинный фиброз
31. Ганглиозидоз
32. Гранюлематоз Вегенера
33. Первичные иммунодефициты
34. Врожденный гиперинсулинизма
35. Гипофосфатазия
36. Хронический гистиоцитоз
37. Ядерная желтуха
38. Общая липодистрофия
39. Синдром кошачьего кика
40. Болезнь Бехчета
41. Болезнь Ильса
42. Болезнь Фарбера
43. Синдром Лея
44. Болезнь Лайма
45. Болезнь (синдром) Такаясу
46. Болезнь Уиппла
47. Спиноцеребеллярная атаксия
48. Тромботические микроангиопатии
49. Врожденные наследственные миопатии
50. Муколипидоз
51. Нарколепсии
52. Нейрофиброматоз
53. Наследственные невропатии
54. Клиническая нейтропения
55. Доброкачественная неакантолитическая пузырчатка
56. Микоскопический полиангиит
57. Узелковый полиартрит
58. Полихондрит
59. Полимиозит
60. Порфирия
61. Псевдоксантома эластическая
62. Истинное преждевременное половое созревание
63. Гипофосфатемический Витамин D-резистентный рахит
64. Боковой амиотрофический склероз Боковой амиотрофический склероз
65. Первичный боковой склероз
66. Туберозный склероз
67. Алкогольный синдром плода
68. Синдром Альстрёма
69. Синдром Ангельмана
70. Синдром Барде-Бидля
71. Аномалия Арнольда – Киари
72. Синдром Беквит-Видемана
73. Синдром Бера
74. Синдром Карпентера
75. Синдром Чарга-Стросса
76. Синдром Когана
77. Синдром Корнелии де Ланге
78. Синдром Итона-Ламберта
79. Синдром Элерса — Данлоса
80. Синдром Фрейзера
81. Синдром Гудпасчера
82. Синдром Кальмана
83. Синдром Клайнфельтера
84. Синдром Леннокса-Гасто
85. Синдром Огучи
86. Синдром Прадера-Вилли
87. Синдром Рассела
88. Синдром Секеля
89. Синдром Стила – Ричардсона
90. Энцефалотригеминальный ангиоматоз
91. Синдром Тернера
92. Болезнь Гиппеля-Линдау
93. Синдром Веста
94. Синдром Вольфрама
95. Синдром Зеллвегер
96. Фетальный гидантоиновый синдром
97. Синдром Мелас
98. Синдром MERRF
99. Врожденный адреногенитальный синдром
100. Синдром хромосомной Анеупладии
101. Синдромы дублирования / дефицита хормосом
Читайте также: