Синдром Тречер Коллинза или Франческетти — Клейна. Челюстно-лицевой дизостоз
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Синдром Тричера Коллинза (TCS ) - это генетическое заболевание, характеризующееся деформациями ушей, глаз, скул и подбородка.. Однако степень поражения человека может варьироваться от легкой до тяжелой. Осложнения могут включать проблемы с дыханием, проблемы со зрением, волчью пасть и потерю слуха. Пострадавшие обычно имеют средний интеллект.
TCS обычно аутосомно-доминантный. Более чем в половине случаев это происходит в результате новой мутации , а не по наследству от родителей человека. Участвующие гены могут включать TCOF1, POLR1C или POLR1D. Диагноз обычно подозревается на основании симптомов и рентгеновских лучей, и потенциальное подтверждение генетическим тестированием.
синдром Тричера Коллинза неизлечим. Симптомы можно лечить с помощью реконструктивной хирургии, слуховых аппаратов, логопеда и других вспомогательных устройств. Продолжительность жизни в целом нормальная. TCS встречается примерно у одного из 50 000 человек. Синдром назван в честь Эдварда Тричера Коллинза, английского хирурга и офтальмолога, которые описали его основные черты в 1900 году.
Содержание
- 1 Признаки и симптомы
- 2 Генетика
- 2.1 TCOF1
- 2.2 Другие мутации
- 3.1 Генетическое консультирование
- 3.2 Пренатальная диагностика
- 3.3 Клинические данные
- 3.4 Рентгенограммы
- 3.5 КТ
- 3.6 Дифференциальный диагноз
- 4.1 Потеря слуха
- 4.2 Психиатрия
Признаки и симптомы
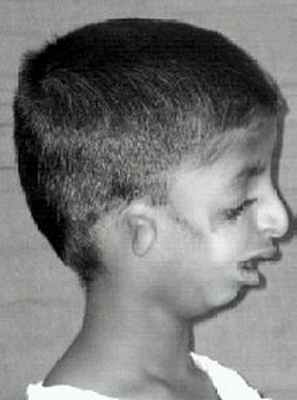
![]()
Тот же ребенок, показанный спереди вверху в информационном окне, теперь виден сбоку, с маленькими ушами и подбородком, который находится далеко позади.
Симптомы у людей с синдромом Тричера Коллинза различаются. У некоторых людей поражение настолько легкое, что их диагноз остается невыявленным, в то время как у других наблюдается умеренное или тяжелое поражение лица и опасные для жизни нарушения дыхательных путей. Большинство признаков ТКС симметричны и распознаются уже при рождении.
Наиболее частым симптомом синдрома Тричера-Коллинза является недоразвитие нижней челюсти и скуловой кости.. Это может сопровождаться втягиванием язычка. Маленькая нижняя челюсть может привести к плохой окклюзии зубов или, в более тяжелых случаях, затрудненному дыханию или глотанию. Дыхательная система ребенка с синдромом Тричера Коллинза является основной проблемой, когда ребенок рождается, и другие проблемы решаются после того, как будут решены респираторные проблемы. Недоразвитие скуловой кости придает щекам запавший вид.
Наружное ухо иногда маленькое, повернуто, деформировано или полностью отсутствует у людей с ТКШ. Также описывается симметричное двустороннее сужение или отсутствие наружного слухового прохода. В большинстве случаев кости среднего уха и полость среднего уха деформированы. Пороки развития внутреннего уха описываются редко. В результате этих аномалий у большинства людей с TCS наблюдается кондуктивная потеря слуха.
У большинства людей также возникают проблемы со зрением, в том числе колобомы (выемки) в нижних веках, частичные или частичные. полное отсутствие ресниц на нижнем веке, веки, наклоненные вниз, опущение верхнего и нижнего века и сужение слезных протоков. Может возникнуть потеря зрения, связанная с косоглазием, аномалиями рефракции и анизометропией. Это также может быть вызвано сильной сухостью глаз, следствием аномалий нижних век и частых глазных инфекций.
Хотя череп неправильной формы не характерен для синдрома Тричера Коллинза, брахицефалия с битемпоральным сужением иногда наблюдается. Расщелина неба также является обычным явлением.
Стоматологические аномалии наблюдаются у 60% пострадавших людей, включая агенез зубов (33%), изменение цвета (помутнение эмали) (20%), неправильное смещение зуба. первые моляры верхней челюсти (13%) и большое расстояние между зубами. В некоторых случаях аномалии зубов в сочетании с гипоплазией нижней челюсти приводят к неправильному прикусу. Это может привести к проблемам с приемом пищи и невозможностью закрыть рот.
Менее распространенные признаки TCS могут усугубить проблемы с дыханием у пострадавшего человека, включая апноэ во сне. Атрезия хоан или стеноз - это сужение или отсутствие хоан, внутреннего отверстия носовых ходов, которое также может наблюдаться. Недоразвитие глотки также может сужать дыхательные пути.
Реже встречаются признаки, связанные с TCS, включая деформации носа, высокое арочное небо, макростомию, преаурикулярные волосы смещение, волчья пасть, гипертелоризм, выемка на верхнем веке и врожденные пороки сердца.
Хотя деформация лица часто связана с задержкой развития и умственной отсталостью, более 95% людей, страдающих TCS, имеют нормальный интеллект. Психологические и социальные проблемы, связанные с лицевой деформацией, могут повлиять на качество жизни людей с СКС.
Генетика
Синдром Тричера Коллинза наследуется по аутосомно-доминантной модели.
Мутации в генах TCOF1, POLR1C или POLR1D могут вызывать синдром Тричера Коллинза. Мутации гена TCOF1 являются наиболее частой причиной заболевания, а мутации генов POLR1C и POLR1D вызывают еще 2% случаев. У людей без идентифицированной мутации в одном из этих генов генетическая причина состояния неизвестна. Гены TCOF1, POLR1C и POLR1D кодируют белки, которые играют важную роль в раннем развитии костей и других тканей лица. Мутации в этих генах снижают производство рРНК, что может вызвать самоуничтожение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица. Непонятно, почему эффекты снижения рРНК ограничиваются развитием лица. Мутации в TCOF1 и POLR1D вызывают аутосомно-доминантную форму Treacher Collins, а мутации в POLR1C вызывают аутосомно-рецессивную форму.
TCOF1
TCOF1 является первичным геном, связанным с TCS; мутация в этом гене обнаруживается у 90-95% людей с TCS. Однако у некоторых людей с типичными симптомами TCS мутации в TCOF1 не обнаружены. Исследование ДНК привело к идентификации мутаций, обнаруженных в TCOF1. Большинство мутаций представляют собой небольшие делеции или вставки, хотя также были идентифицированы сайт сплайсинга и миссенс-мутации.
Анализ мутаций выявил более 100 болезнетворных мутаций в TCOF1, которые в основном являются семейно-специфическими мутациями. На единственную рецидивирующую мутацию приходится около 17% случаев.
TCOF1 обнаруживается на 5-й хромосоме в области 5q32. Он кодирует относительно простой ядрышковый белок, называемый патока, который, как считается, участвует в сборке рибосом. Мутации в TCOF1 приводят к гаплонедостаточности белка патоки. Гаплонедостаточность возникает, когда диплоидный организм имеет только одну функциональную копию гена, потому что другая копия инактивирована мутацией. Одна нормальная копия гена не производит достаточного количества белка, что вызывает болезнь. Гаплонедостаточность белка патоки приводит к истощению предшественника клеток нервного гребня, что приводит к уменьшению количества клеток гребня, мигрирующих в первую и вторую глоточные дуги. Эти клетки играют важную роль в развитии черепно-лицевого внешнего вида, и потеря одной копии патоки влияет на способность клеток формировать кости и ткани лица.
Другие мутации
Мутации POLR1C и POLR1D ответственны за меньшую часть случаев Treacher Collins. POLR1C находится на хромосоме 6 в положении 6q21.2, а POLR1D находится на хромосоме 13 в положении 13q12.2. Эти гены кодируют белковые субъединицы, общие для РНК-полимеразы I и III. Обе эти полимеразы важны для биогенеза рибосом.
Диагноз
Генетическое консультирование
TCS наследуется аутосомно-доминантным способом и пенетрантностью пораженный ген почти завершен. Однако некоторые недавние исследования описали некоторые редкие случаи, когда пенетрантность в TCS не была полной. Причинами могут быть переменная экспрессия, неполная пенетрантность или мозаицизм зародышевой линии. Только 40% мутаций передаются по наследству. Остальные 60% являются результатом мутации de novo, когда ребенок имеет новую мутацию в ответственном гене и не унаследовал ее ни от одного из родителей. В исходе болезни возникает межсемейная и внутрисемейная изменчивость. Это говорит о том, что при рождении пораженного ребенка важно обследовать родителей, чтобы определить, присутствует ли пораженный ген, поскольку у родителя может быть легкая форма заболевания, которая не была диагностирована. В этом случае риск рождения еще одного больного ребенка составляет 50%. Если у родителей нет пораженного гена, риск рецидива низок. В следующих поколениях тяжесть клинических симптомов увеличивается.
Пренатальная диагностика
Мутации в основных генах, ответственных за TCS, могут быть обнаружены с помощью взятия проб ворсинок хориона или амниоцентеза. Эти методы не позволяют обнаружить редкие мутации. Ультрасонография может использоваться для выявления черепно-лицевых аномалий на более поздних сроках беременности, но может не выявить более легкие случаи.
Клинические данные
TCS часто сначала подозревают при наличии характерных симптомов, наблюдаемых во время физического осмотра. Однако клинические проявления СТК могут напоминать другие заболевания, что затрудняет диагностику. Классификация OMENS была разработана как комплексный и поэтапный подход к дифференциации заболеваний. Этот акроним описывает пять различных проявлений дисморфологии, а именно орбитальную асимметрию, гипоплазию нижней челюсти, деформацию ушной раковины, развитие нервов и заболевание мягких тканей.
- O0: нормальный размер орбиты, положение
- O1: аномальный размер орбиты
- O2: аномальное положение орбиты
- O3: аномальный размер и положение орбиты
- M0: нормальная нижняя челюсть
- M1: маленькая нижняя челюсть и гленоид ямка с короткой ветвью
- M2: ветвь короткая и неправильной формы
- 2A: гленоидная ямка в анатомически приемлемом положении
- 2B: височно-нижнечелюстной сустав снизу (ВНЧС), медиально, смещенный кпереди, с сильно гипоплазированным мыщелком
- M3: полное отсутствие ветви, суставной ямки и височно-нижнечелюстного сустава
- E0: нормальное ухо
- E1: незначительная гипоплазия и купирование всех структур
- E2: Отсутствие наружного слухового прохода с различной гипоплазией ушной раковины
- E3: Неправильное положение дольки с отсутствием ушной раковины, дольчатый остаток обычно нижний смещенный кпереди
- N0: нет лицевой нерв вовлечение
- N1: поражение верхнего лицевого нерва (височные или скуловые ветви)
- N2: Поражение нижнего лицевого нерва (буккального, нижнечелюстного или шейного)
- N3: поражены все ветви
- S0: нет дефицита мягких тканей или мышц
- S1: минимальное количество тканей или мышц дефицит
- S2: умеренный тканевый или мышечный дефицит
- S3: тяжелый тканевый или мышечный дефицит
Рентгенограммы
Для подтверждения диагноза в TCS используется несколько методов. 99>
ортопантомограмма (OPG) - это панорамный рентгеновский снимок верхней и нижней челюсти. Он показывает двухмерное изображение от уха до уха. В частности, OPG способствует точному послеоперационному наблюдению и мониторингу роста костей при лечении с использованием моно- или двойного дистрактора. Таким образом, некоторые особенности TCS можно увидеть на OPG, но используются более совершенные методы, позволяющие охватить весь спектр аномалий TCS вместо того, чтобы показывать только аномалии челюсти.
Другой метод радиографической оценки - получение рентгеновского изображения всей головы. Боковая цефалометрическая рентгенограмма в TCS показывает гипоплазию лицевых костей, таких как скуловая кость, нижняя челюсть и сосцевидный отросток.
Наконец, затылочные рентгенограммы используются для обнаружения гипоплазии или нарушения целостности скуловой дуги.
КТ височной кости с использованием тонких срезов позволяет диагностировать степень стеноза и атрезии наружного слухового прохода, состояние полости среднего уха, отсутствие или диспластические и рудиментарные косточки, или аномалии внутреннего уха, такие как недостаточная улитка. Двух- и трехмерные КТ-реконструкции с VRT и покрытием костей и кожи полезны для более точной постановки и трехмерного планирования реконструктивной хирургии нижней челюсти и наружного уха.
Дифференциальный диагноз
Другие болезни имеют сходные характеристики с синдромом Тричера Коллинза. В дифференциальной диагностике следует учитывать акрофациальные дизостозы. Внешний вид лица напоминает синдром Тричера Коллинза, но у этих людей возникают дополнительные аномалии конечностей. Примерами этих заболеваний являются синдром Нагера и синдром Миллера.
При дифференциальной диагностике также следует учитывать окулоаурикуловертебральный спектр. Примером является гемифациальная микросомия, которая в первую очередь влияет на развитие уха, рта и нижней челюсти. Эта аномалия может возникать двусторонне. Еще одно заболевание, относящееся к этому спектру, - синдром Гольденхара, который включает в себя позвоночные аномалии, эпибульбарные дермоиды и деформации лица.
Лечение
Лечение людей с TCS может включать вмешательство профессионалов из разных дисциплин. В первую очередь заботятся о дыхании и кормлении из-за гипоплазии нижней челюсти и обструкции гортани языком. Иногда им может потребоваться трахеостомия для поддержания проходимости дыхательных путей и гастростомия для обеспечения адекватного потребления калорий и защиты дыхательных путей. Корректирующая хирургия лица проводится в определенный возраст, в зависимости от состояния развития.
Обзор настоящих рекомендаций:
- Если присутствует волчья пасть, восстановление обычно требует место в возрасте 9–12 месяцев. Перед операцией необходима полисомнография с установленной небной пластиной. Это может предсказать послеоперационную ситуацию и дает представление о вероятности наличия апноэ во сне (СОАС) после операции.
- Для лечения потери слуха используется усиление костной проводимости, логопедия и образовательное вмешательство во избежание языковых / речевых проблем. слуховой аппарат с костной фиксацией - альтернатива для людей с аномалиями уха.
- Скуловая и орбитальная реконструкция выполняется, когда краниоорбитозигоматическая кость полностью сформирована, обычно в возрасте от 5 до 5 лет. 7 лет. У детей чаще всего используют аутотрансплантат. В сочетании с этой трансплантацией можно использовать липофилинг в периорбитальной области для получения оптимального результата реконструкции. Реконструкция нижнего века колобома включает использование кожно-мышечного лоскута, который приподнимается и таким образом закрывает дефект века.
- Реконструкция наружного уха обычно выполняется, когда человеку не менее восемь лет. Иногда можно лечить наружный слуховой проход или среднее ухо.
- Оптимальный возраст для проведения реконструкции челюстно-нижнечелюстной кости является спорным; по состоянию на 2004 год использовалась эта классификация:
- Тип I (легкая форма) и Тип IIa (умеренная) 13–16 лет
- Тип IIb (порок развития средней и тяжелой степени) при зрелости скелета
- Тип III (тяжелый) 6–10 лет
- Когда зубы режутся, зубы должны находиться под наблюдением ортодонта, чтобы убедиться в отсутствии аномалий. При обнаружении таких аномалий, как вывих или чрезмерное разрастание зубов, соответствующие меры могут быть предприняты как можно скорее.
- Ортогнатическое лечение обычно проводится после 16 лет; к этому моменту все зубы встали на свои места, а челюсти и протезы стали зрелыми. При обнаружении СОАС уровень обструкции определяется с помощью эндоскопии верхних дыхательных путей. Выдвижение нижней челюсти может быть эффективным способом улучшить как дыхание, так и эстетику, в то время как пластика подбородка только восстанавливает его профиль.
- Если реконструкция носа необходима, она обычно выполняется после ортогнатической операции и в возрасте старше 18 лет..
- Контур мягких тканей лица обычно требует коррекции в более позднем возрасте из-за зрелости лицевого скелета. Использование микрохирургических методов, таких как перенос свободного лоскута, улучшило коррекцию контуров мягких тканей лица. Еще один метод улучшения контуров мягких тканей лица - липофилинг. Например, липофилинг используется для реконструкции век.
Потеря слуха
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха обычно двусторонняя с кондуктивной потерей около 50-70 дБ. Даже в случаях с нормальными ушными раковинами и открытыми наружными слуховыми проходами цепочка слуховых косточек часто деформируется.
Попытки хирургической реконструкции наружного слухового прохода и улучшения слуха у детей с СКС не дали положительных результатов.
Слуховая реабилитация с помощью слуховых аппаратов с костной фиксацией (BAHA) или обычных средств костной проводимости оказалась предпочтительнее хирургической реконструкции.
Психиатрические
Заболевание может быть ассоциировано с рядом психологических симптомов, включая тревогу, депрессию, социальную фобию и беспокойство по поводу образа тела; люди также могут подвергаться дискриминации, издевательствам и обзываниям, особенно в молодом возрасте. Эти вопросы должны учитывать многопрофильная команда и родительская поддержка.
Эпидемиология
TCS встречается примерно у одного из 50 000 новорожденных в Европе. По оценкам, во всем мире он встречается от одного из 10 000 до 1 из 50 000 рождений.
История
Синдром назван в честь Эдварда Тричера Коллинза (1862–1932), англичанин хирург и офтальмолог, которые описали его основные черты в 1900 году. В 1949 году Адольф Франческетти и Дэвид Кляйн описали то же заболевание на основании своих собственных наблюдений как нижнечелюстно-лицевой дизостоз.. Термин нижнечелюстно-лицевой дизостоз используется для описания клинических проявлений.
Культура
Статья в New York Times, июль 1977 г. , перепечатанная в многочисленных газетах по всей стране в течение следующих недель. эта болезнь впервые привлекла внимание многих людей.
Беспорядок был показан в шоу Nip / Tuck, в эпизоде «Blu Mondae ». TLC «Рожденные без лица» с Джулианой Ветмор, которая родилась с самым тяжелым случаем в истории болезни этого синдрома, у которой отсутствуют 30-40% костей лица.
В 2010 году BBC Three документальный фильм Люби меня, люби мое лицо, освещал дело человека, Джоно Ланкастера, с этим состоянием. В 2011 году BBC Three вернулся к Джоно, чтобы осветить его и его партнершу Лору стремление создать семью в фильме «Так что, если мой ребенок родился как я?», Который впервые вышел в эфир в рамках сезона программ BBC Three о воспитании детей. Первый фильм был показан на BBC One незадолго до первой трансляции второго фильма на BBC Three. Третий фильм Ланкастера BBC Three, «В поисках семьи» на Facebook, в котором рассматривается усыновление, вышел в эфир в 2011 году.
В Wonder, детский роман, главный герой - ребенок с синдромом Тричера Коллинза. В ноябре 2017 года вышла экранизация 2017 года с Джулией Робертс, Оуэном Уилсоном и Джейкобом Тремблеем. Элисон Мидстокк, актриса и модель с этим заболеванием.
Синдром Тричера-Коллинза-Франческетти (мандибуло-фациальный дизостоз). Поиск мутаций в гене TCOF1, м. (Treacher-Collins Syndrome, Franceschetti-Klein Syndrome, Mandibulofacial Dysostosis without Limb Anomalies, Gene TCOF1, Mut.)
Синдром Тричер – Коллинза - Франческетти (мандибулофациальный дизостоз, TCOF, OMIM154500) - аутосомно-доминантное заболевание, характеризующееся нарушением черепно-лицевого развития. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное нёбо или расщелина нёба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу: крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. При тяжёлой патологии микрогнатия может вытеснять язык у новорождённых, создавая опасную преграду в ротоглотке для дыхания.
Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в генеTCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма). Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. Возможна прямая молекулярно-генетическая диагностика этого синдрома, которая заключается в выявлении изменений нуклеотидной последовательности в гене TCOF1 методом прямого автоматического секвенирования.
Аутосомно-доминантный. Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни).
Мутацию в гене TCOF1 можно выявить в 78%-93% случаев, в 8% выявляют мутации в генах POLR1C или POLR1D.
Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. У больных весьма характерное лицо. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное небо или расщелина неба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. Порок развития уха заключается в деформации ушной раковины, отсутствии костного отдела наружного слухового прохода, недоразвитии барабанной полости и слуховых косточек. Отмечаются также гипоплазия больших пальцев лучевой и локтевой костей, расщелины неба. Тугоухость смешанного характера с одновременным поражением звукопроведения и звуковосприятия.
Интеллект, как правило, не страдает. Трудности с дыханием и питанием могут возникнуть в первые годы из-за стеноза верхних дыхательных путей и ограниченного открытия рта.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Специальной подготовки к исследованию не требуется.
- анкета генетического исследования *;
- направительный бланк;
- информированное согласие.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Республики Беларусь.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Типичная клиническая картина.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
синдром I-II жаберных дуг, гемифациальная микросомия, синдром Гольденхара, синдром Пьера Робена, синдромы Нагера.
- Мутация не выявлена.
- Мутация выявлена в гетерозиготном состоянии.
- Мутация выявлена в гомозиготном состоянии.
- Мутация выявлена в компаунд –гетерозиготном состоянии.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Сдать анализ "Синдром Тричера-Коллинза-Франческетти (мандибуло-фациальный дизостоз). Поиск мутаций в гене TCOF1, м. (Treacher-Collins Syndrome, Franceschetti-Klein Syndrome, Mandibulofacial Dysostosis without Limb Anomalies, Gene TCOF1, Mut.)" вы можете в Минске и других городах Республики Беларусь. Обратите внимание, что цена анализа, стоимость процедуры взятия биоматериала, методы и сроки выполнения исследований в региональных медицинских офисах могут отличаться
Синдром Тречер Коллинза или Франческетти — Клейна. Челюстно-лицевой дизостоз
Синдром Тречер Коллинза или Франческетти — Клейна. Челюстно-лицевой дизостоз
Хотя синдром был, возможно, впервые описан Thomson (1846— 1947), заявка на это открытие обычно отдается Berry или особенно Treacher Collins (имя обычно часто ошибочно связывают дефисом), который описал существенные компоненты синдрома. Franceschelti и Klein опубликовали обширный обзор, посвященный этому синдрому, и дали ему название челюстно-лнцевого дизостоза. Rogers произвел разносторонний анализ случаев. Всего опубликовано более чем 250 случаев.
Клинические данные. Данные осмотра. Внешний вид лица характерен. Косой опущенный вниз разрез глаз, запавшие кости щек, деформированная ушная раковина, отступающий назад подбородок (ретрогнатия) и большой, похожий на рыбий, рот так хорошо представляют клиническую картину болезни, что делают ее незабываемой. Другой необычной чертой является своеобразный рост волос в виде языка, распространяющегося в направлении щек (Gorlin et al.).
Несмотря на то что разрез глаз имеет косую, антимонголоидную форму, зрение обычно нормальное. Часто (примерно у 75% больных) имеется колобома в наружной трети нижнего века. Почти у 50% больных отмечается недостаток ресниц медиальнее колобомы.
Носолобный угол обычно закрыт, а переносье приподнято. Из-за недостаточного развития скуловых костей нос выглядит крупным. Ноздри нередко узки, а хрящ носа гипопластичен.Нижняя челюсть почти всегда гипопластична, угол ее более тупой, чем в норме, а рама недоразвита. Под поверхностью тела нижней челюсти часто имеется резко выраженная впадина. Более чем у 40% больных отмечается высокое или расщепленное небо, а также недостаточное смыкание зубов.
Орган слуха. Ушная раковина часто деформирована, сморщена спереди или смещена. Наружный слуховой проход, когда он имеется, узкий и косой (Harrison). Около 85% больных имеют деформированную ушную раковину и более чем у 1/3 больных отсутствует наружный слуховой проход или отмечается дефект слуховых косточек с проводящей глухотой (Stovin et al.). Отмечен также нейросенсорный компонент глухоты (Kittel, Fleischer-Peters, Partsch, Hulse).
Рентгенологические исследования показали наличие склероза среднего и изредка внутреннего уха с плохо очерченными структурами. Слуховые косточки, а также улитка и вестибулярный аппарат могут отсутствовать или быть грубо деформированными (McKenzie, Craig, Pavsek, Herberts).
Хирургическое исследование выявило такие аномалии, как фиксацию молоточка, синостоз и уродство молоточка и наковальни, отсутствие стремени и овального окна, отсутствие сухожилия m. stapedius, деформацию наковальни и стремени, отсутствие наковальни, анкилоз основания стремени, костный мостик в канале лицевого нерва, а также полное отсутствие среднего уха и надбарабанного пространства. Полость среднего уха может быть заполнена соединительной тканью (Altmann, Plester, Holborow, Keerl). Plester, Keerl и Edwards обнаружили монолитное стремя и истончение длинного отростка наковальни.
![челюстно-лицевой дизостоз]()
Почти у 25% больных была обнаружена патология лабиринта (Stovin, Herberts). Между трагусом и углом рта могут встречаться внеушные бугорки или слепые свищи.
Вестибулярная система. Результаты исследований вестибулярной системы не опубликованы.Лабораторные данные. Рентгенологически нижний край глазницы дефектен, а полость глазницы овальной формы с наклоненной вниз и наружу крышей. Тело скуловой кости может вовсе отсутствовать, по чаще, однако, оно значительно и симметрично недоразвито и не соединено со скуловыми дугами (нет височной ветви и фактически отсутствует верхнечелюстная ветвь). Сосцевидный отросток безвоздушен и. часто склерозирован. Параназальные синусы очень малы и могут полностью отсутствовать.
Томография височных костей показала атрезию наружного слухового прохода, значительное уменьшение размера барабанной полости, частую недостаточность верхнего отдела среднего уха, утолщение ушной капсулы, расположение дна средней ямки ниже уровня наружного полукружного канала и неправильное направление лицевого нерва (Terrahe).
Наследственность. Синдром наследуется по аутосомно-доминантному типу с неполной пенетрантностью и варьирующей экспрессивностью. Создается впечатление, что ген обладает летальным эффектом, так как в семьях часто наблюдаются выкидыши или ранняя постнатальиая смерть детей.
Диагноз. От челюстно-лицевого дизостоза необходимо отличать глазо-ушно-позвоночный синдром (окуло-аурикуло-вертебральный синдром, синдром Гольденхара, гемифациальная микросомия). Характерным для синдрома Гольденхара является асимметрия лица, односторонняя гипоплазия рамы нижней челюсти, деформированная ушная раковина. Менее часто наблюдаются эпибульбарная дермоидная киста, колобома верхнего века, ушные бугорки, макростомия, односторонний парез лицевого нерва и аномалии позвонков (Gorlin et al.). Наследование, вероятно, мультифакторнальное.
Лечение. Совместные усилия должны быть направлены на коррекцию микротии, гипоплазии нижней челюсти, песмыкаиия зубов, гипоплазии скуловых костей. К улучшению слуха обычно ведет тимпанотомия с введением слухового протеза.
Прогноз. Глухота не прогрессирует.Выводы. Характеристика этого синдрома включает: 1) аутосомно-доминантное наследование с варьирующей экспрессивностью; 2) гипоплазию скуловых костей, в результате чего возникает антимонголоидный разрез глаз; 3) колобому нижнего века п отсутствие ресниц медиальнее колобомы; 4) гипоплазию нижней челюсти; 5) деформацию ушной раковины, наружного слухового прохода и структур среднего уха; 6) проводящую глухоту.
Синдром Тричера-Коллинза ( Мандибулофациальный дизостоз , Синдром Тричера Коллинза-Франческетти , Челюстно-лицевой дизостоз )
Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
МКБ-10
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
Причины
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 - №3.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма
![]()
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
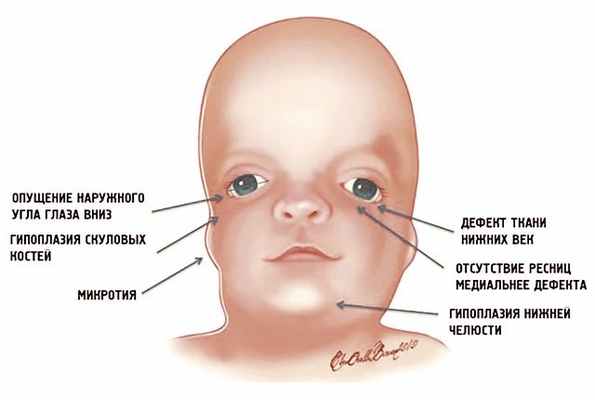
![Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза]()
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
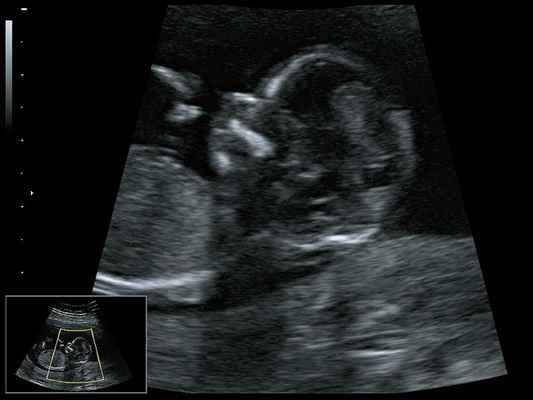
![Микрогнатия - сагиттальный скан в 2D, беременность 13 нед]()
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Читайте также: