Синдром Brugada. Генетические основы синдрома Бругада
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Синдром Бругады является генетическим заболеванием , расстройство характеризуется ИМПСТ сегментом ST (в) на ведущую правых прекордиальных V1, V2 и V3, и блок аспект правой ветви в электрокардиограмме , связанную с высоким риском желудочковых аритмий , которые могут вызвать обморок и внезапная смерть, структурно нормальное сердце. Передача аутосомно-доминантная, пенетрантность варьирует. Генетические мутации приводят к нарушениям ионных каналов. Средний возраст первого клинического эпизода - 40 лет, с сильным преобладанием мужчин. По оценкам, распространенность составляет около 1/1000 в азиатских странах, вероятно, ниже в других странах. Прогноз для пациентов с симптомами тяжелый, и внезапную смерть можно предотвратить, установив автоматический имплантируемый дефибриллятор .
Резюме
Исторический
Этот синдром был впервые описан в 1992 году братьями Педро и Хосеп Бругада.
Этиология
Синдром Бругада - это натриевая каннелопатия .
Одной из причин является мутация в с SCN5A гена , расположенного на хромосоме 3 . В этом гене было идентифицировано почти 300 различных мутаций, но эти мутации встречаются только в одном из пяти случаев Brugada.
Это аутосомно-доминантное заболевание с низкой пенетрантностью (некоторые носители мутации проявляют признаки болезни).
SCN10A , HEY2 , GPDL1 гены (ген фермента глицерол-3-фосфат дегидрогеназы 1 т.п.) или CACNA1C могут быть вовлечены. У других пораженных пациентов без мутации SCN5A заболевание присутствует, но ответственный ген не идентифицируется в трех четвертях случаев. Высказывается гипотеза о многофакторном происхождении. Генетическое типирование, похоже, пока не имеет прогностического интереса. Это кажется менее верным с развитием исследований.
На электрофизиологическом уровне, по-видимому, существует неоднородность задержек проводимости в правом желудочке, которая может быть субстратом для запуска и сохранения ритмических нарушений. Это может быть связано с замедленной проводимостью на уровне промывочной камеры правого желудочка и с низким напряжением, с фиброзом и дефицитом сообщающихся соединений в этом месте. Воспалительный механизм на этом уровне часто ассоциируется.
Эпидемиология
По оценкам, частота составляет 1 из 1000 в азиатских популяциях, где синдром внезапной смерти во сне является обычным явлением; это вторая по значимости причина внезапной смерти молодых людей в Азии после дорожно-транспортных происшествий.
Средний возраст постановки диагноза или внезапной смерти составляет 40 плюс-минус 22 года, при этом крайний возраст составляет от двух дней жизни до более 80 лет.
Кажется, это чаще встречается у мужчин, у последних более серьезные формы.
Описание
Диагноз синдрома Бругада основан на ассоциации отклонений на ЭКГ (подъем сегмента ST) и таких событий, как обморок или внезапная смерть. Все чаще кардиологи вынуждены прокомментировать реальность этого диагноза у бессимптомных пациентов, у которых на ЭКГ только аномальные проявления (подъем сегмента ST).
На практике необходимо отличать диагностированный синдром Бругада от простого появления Бругада на электрокардиограмме, которое часто обнаруживается случайно, и риски которого очень изменчивы (но в среднем ниже).
Диагностический
Элементы, участвующие в диагностике:
- семейный анамнез синдрома Бругада, внезапная смерть;
- личная история внезапной реанимационной смерти;
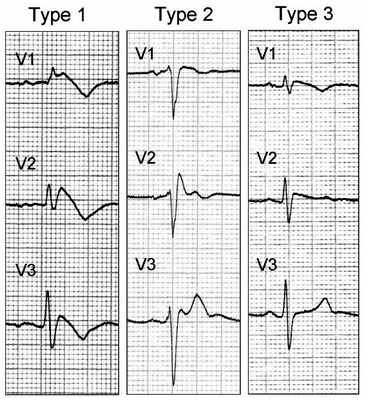
- на электрокардиограмме: аспект блокады правой ножки пучка Гиса, связанный с подъемом сегмента ST и аномалиями зубца Т в правых прекардиальных отведениях (от V1 до V3). Описаны три типа отклонений ЭКГ:
- тип 1: куполообразная элевация сегмента ST,
- тип 2: подъем сегмента ST в виде конского седла,
- тип 3: тот же рисунок на электрокардиограмме, что и тип 2, но уплощенный,
Разделение на три типа по электрокардиограмме иногда бывает затруднительным, потому что последний может меняться изо дня в день у одного и того же человека, переходя от одного типа к другому. Повышение может увеличиваться во время фазы восстановления во время стресс-теста в трети случаев, и тогда это будет уничижительным с точки зрения прогноза. Кроме того, тип 1 встречается чаще, если электроды располагаются на груди на межреберье выше, чем обычно. Могут быть связаны и другие электрические аномалии: таким образом, ранняя реполяризация (вогнутое смещение вершины сегмента ST) на нижних территориях наблюдается в одном из 10 случаев и может быть уничижительным критерием.
![]()
Поддерживается
Некоторые рекомендации по лечению Brugada были опубликованы в 2005 году. Другие, американские и европейские, о нарушениях ритма генетического происхождения были опубликованы в 2013 году.
Во всех случаях следует избегать или лечить любые известные триггерные обстоятельства Brugada типа I. В частности, следует незамедлительно лечить любую лихорадку.
Лечение зависит от типа аномалии ЭКГ, результата фармакологического теста, семейного анамнеза внезапной смерти, симптомов и нарушений ритма, представленных пациентом. В случае полного и типичного синдрома Бругада с в анамнезе восстановленной остановкой сердца и отсутствием диагностических сомнений показание к установке автоматического имплантируемого дефибриллятора (ИКД) является общепринятым. Это показание более обсуждается в других случаях (в частности, изолированной аномалии электрокардиограммы), при этом риск внезапной смерти значительно ниже. Однако он не равен нулю, и проблема, по сути, основана на стратификации этого риска.
В начале 2000-х годов велись научные дебаты, которые до сих пор не решены, что делать в случае спонтанного электрокардиографического появления типа 1 у бессимптомного пациента. Ранее было рекомендовано электрофизиологическое исследование с протоколом запрограммированной кардиостимуляции правого желудочка, чтобы найти триггерное нарушение желудочкового ритма, первые данные, казалось бы, показывают, что это был уничижительный критерий, но это не всегда подтверждалось более свежими данными и интересами. остается дискуссионным. Точно так же семейный анамнез внезапной смерти , по-видимому, не увеличивает этот риск у бессимптомных лиц. Терапевтическая «санкция» варьируется в зависимости от баланса между преимуществами и рисками, от отсутствия конкретной рекомендации (субъект, личный риск которого рассматривается как риск для здорового населения в целом) до внедрения имплантируемого автоматического дефибриллятора. который обеспечит электрошок ( дефибрилляцию ) в случае нарушения желудочкового ритма.
Медикаментозного лечения для предотвращения желудочковых аритмий при синдроме Бругада не существует. Хинидин используется некоторыми, пока эмпирически.
Радиочастотная абляция , по эпикардиальному пути из области в выносящем тракте правого желудочка, позволяет исчезновению аномалии на ЭКГ , но ее полезность в предотвращении внезапной смерти еще предстоит установить.
Генетическое консультирование
Имеющиеся в настоящее время данные о механизме этого заболевания говорят в пользу большого количества случаев мутаций рецепторов каналов, в частности натриевых каналов в клетках миокарда, поэтому иногда можно попытаться найти семейные мутации, когда есть аргументы в пользу. несколько семейных дел. Однако это исследование вызывает сомнения: на практике наличие мутации, похоже, не коррелирует с повышенным риском серьезных событий.
Синдром Brugada. Генетические основы синдрома Бругада
Синдром Brugada. Генетические основы синдрома Бругада
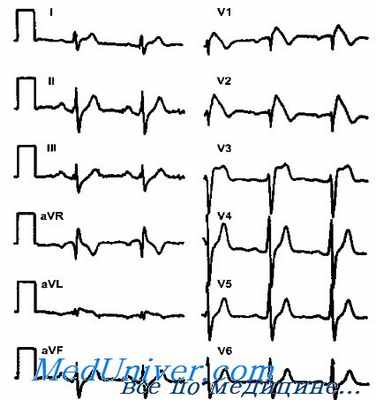
В 1992 г. Brugada P. и Brugada R. описали пациентов с подъемом сегмента ST в правых прекорлиальных отведениях (V1-V3) (в отсутствие острого коронарного синдрома), с блокадой правой ножки пучка Гиса, со склонностью к желудочковым тахиаритмиям при структурно нормальном сердце. Сообщалось о семейном наследовании по аутосомно-доминантному типу, предполагая генетическое происхождение болезни. Эта болезнь в настоящее время носит название «синдром Brugada». Частота ее распространения неизвестна, но болезнь чаще встречается в дальневосточных странах.
Типичная аритмия при синдроме Brugada — быстрая полиморфная ЖТ, которая часто переходит в ФЖ; задокументирован также переход в фибрилляцию предсердий. Несмотря на то что синдром Brugada — генетически обусловленное заболевание, клинические проявления (обморок или остановка сердца) в детском возрасте редки и появляются в третьей и четвертой декадах жизни в соотношениях 8 : 1 (мужчины/женщины). Причины таких возрастных и тендерных различий неизвестны. Сердечные события развиваются во время сна или в покое.
Лихорадка, трициклические антидепрессанты и употребление кокаина могут быть пусковыми механизмами сердечных событий у некоторых пациентов.Диагноз синдрома Brugada затруднен из-за преходящих изменений па электрокардиограмме. Скрытые формы могут быть обнаружены во время пробы с антиаритмиками класса 1С: аймалином (1 мг/кг), флекаинидом (2 мг/кг) или прокаинамидом (15 мг/кг). Автономная модуляция нервной системы может изменить проявления синдрома: внутривенное введение изопротеренола уменьшает ЭКГ-признаки синдрома Brugada, а анетилхолина увеличивает.
Эта особенность согласуется с тем, что сердечные события при синдроме Brugada наступают главным образом и покое или во время сна. Диагностические критерии синдрома Brugada уточнялись в течение нескольких лет. Выли установлены три типа ЭКГ, однако диагностическим критерием считается только «выпуклая» элевация сегмента ST (тип I). Опасные для жизни сердечные события могут развиться у пациентов с ЭКГ типа «спинка седла» (тип II) с генетически подтвержденным диагнозом.
![синдром бругада]()
Генетические основы синдрома Бругада
Первый ген был идентифицирован в 1998 г. как сердечный теп ионного канала натрия (SCN5A). Этот же теп ответственен за генетический вариант LQT3. Хотя синдром Brugada и LQT3 вызваны мутациями в одном и том же гене, существует противоположный эффект этих мутации на ионный ноток натрия: «потеря функции» при синдроме Brugada и усиление функции при LQT3.
Интересно, что о пересечении фенотипов между LQT3 и синдромом Brugada сообщили независимые исследователи, которые подчеркнули функциональную сложность сердечной «болезни ионного канала». Недавно была обнаружена связь мутации SCN5A с ДКМП, что предполагает возникновение структурных нарушений сердца как прямого следствия генетически измененных электрофизиологических свойств кардиомиоцитов. Особенно это относится к пациентам с синдромом Brugada, поскольку наличие структурных нарушений является частью фенотипа.
Было высказано предположение, что вирусная инфекция может служить частью пускового механизма, который сопровождает развитие структурных нарушении у пациентов с синдромом Brugada с мутацией SCN5A или без нее.В связи с этим при клиническом ведении пациентов необходимо осторожно оценивать ЭКГ у всех пациентов с синдромом Brugada.
Недавно о втором генетическом варианте синдрома Brugada сообщили на примере одной большой семьи с мутацией гена GPD1-L, кодирующего белок, похожий на глицерол-3-фосфат-дсгидрогеназу-1. Функция этого белка в сердце полностью не изучена, по предварительные экспериментальные данные показывают, что мутация гена GPD1-L. уменьшает экспрессию ионного потока натрия.Несмотря на то что клиническое значение генетического тестирования при синдроме Brugada ограничено, в случаях, когда оно доступно, эта информация полезна для идентификации «немых носителей» и для доклинической диагностики у членов семьи пробанда.
Синдром Бругада
Синдром Бругада (СБ) – это генетическое заболевание сердца, сопряженное с повышенным риском внезапной смерти.
Обычно заболевание развивается в зрелом возрасте, в среднем около 40 лет, и, по оценкам, является причиной не менее 4% всех случаев внезапной смерти, среди которых не менее 20% приходится на пациентов со структурно нормальным сердцем.
В большинстве случаев СБ диагностируют молодым совершеннолетним пациентам, чаще всего мужского пола, после внезапной смерти члена семьи или при появлении таких симптомов, как обморок или остановка сердца. СБ имеет характерную электрокардиографическую структуру, представленную типичной элевацией сегмента ST в правых прекордиальных электрокардиографических отведениях (от V1 до V3), называемой паттерном Бругада 1 типа. Если электрокардиографические признаки предполагаются, но не диагностированы (паттерн Бругада 2 или 3 типа), для подтверждения или исключения диагноза может потребоваться проведение теста с антиаритмическими препаратами I класса (например, аймалином или флекаинидом), которые обнаруживают диагностический (тип 1) паттерн Бругада.
Обычно пациенты с СБ имеют структурно нормальное сердце, хотя магнитно-резонансная томография выявила легкие структурные аномалии правого желудочка у подгруппы пациентов.
Это заболевание, по-видимому, связано с нарушением работы одного или нескольких ионных каналов - структур, обеспечивающих транзит этих ионов (натрия, калия, магния и кальция) через поверхность клетки. Первым генетическим изменением, выявленным при СБ, было изменение гена SCN5A, кодирующего натриевый канал. Впоследствии с СБ были связаны многочисленные и новые генетические аномалии; описано более 300 мутаций.
Каковы симптомы заболевания?
Характерной особенностью СБ является чрезвычайная вариабельность клинической картины.
К сожалению, у значительного процента пациентов первым клиническим проявлением заболевания является внезапная смерть, вызванная летальными желудочковыми аритмиями. Последнее катастрофическое явление подчеркивает сложность лечения заболевания в плане прогнозирования того, какие люди будут подвержены наибольшему риску, поскольку до этого явления у подавляющего большинства пациентов не наблюдается каких-либо тревожных симптомов.
Заболевание может протекать абсолютно бессимптомно или проявляться симптомами, связанными с сердечной аритмией, с эпизодами обморока (т.е. потерей сознания), приступами учащенного сердцебиения или повышения частоты сердечных сокращений. Иногда у пациентов могут наблюдаться судороги и симптомы, которые можно спутать с эпилепсией. Зачастую симптомы развиваются в состоянии покоя, на этапе восстановления после физической нагрузки или во время ночного сна.
- учащенное сердцебиение
- одышка
- усталость
- обморок
Как диагностируется?
Своевременная диагностика имеет решающее значение при этом синдроме, поскольку пациент может подвергаться риску развития потенциально летальных аритмий. При подозрении на синдром Бругада необходимо собрать семейный анамнез и электрокардиографическую трассировку, а также провести другие кардиологические исследования, включая эхокардиограмму (чтобы исключить наличие какого-либо заболевания сердца).
Подтверждение диагноза синдрома Бругада возможно при наличии спонтанного паттерна 1-го типа.
Однако в большинстве случаев картина ЭКГ позволяет предположить, но не диагностировать СБ (паттерн 2 или 3 типа), и может потребоваться фармакологический тест с аймалином, который у предрасположенных пациентов вызывает появление паттерна Бругада 1 типа и, следовательно, позволяет исключить или подтвердить диагноз синдрома Бругада.
В случае положительного фармакологического теста может потребоваться эндокавитальное электрофизиологическое исследование (ЭЭС), чтобы проверить возможную уязвимость пациента к потенциально злокачественным желудочковым аритмиям.
Рекомендуемая диагностика
- Электрокардиограмма (ЭКГ)
- ЭКГ по Холтеру
- Стресс-тест
- Эхокардиограмма
- КардиоТелефон
- Пассивная ортостатическая проба (тилт-тест)
Как лечится?
После постановки диагноза СБ терапевтический подход зависит от степени риска пациента. Синдром Бругада не имеет определенного клинического развития, поэтому у пациента могут появляться новые клинические элементы, указывающие на возможное изменение степени риска. Поэтому целесообразно проводить проверки каждые шесть месяцев.
В настоящее время терапевтические варианты, доступные для лечения синдрома Бругада, относятся в основном к трем типам:
- Имплантируемые сердечные дефибрилляторы (ИСД, внутриполостные или подкожные) или имплантируемые кардиомониторы (подкожный ИКМ или петлевой регистратор)
- Транскатетерная эпикардиальная абляция аритмического субстрата
- Медикаментозная терапия специфическими антиаритмическими препаратами (хинидин)
В нашем центре мы разработали подход, основанный на клинических характеристиках каждого конкретного человека, на базе нашего обширного и длительного клинического опыта, включающего тысячи случаев с продолжительным последующим наблюдением.
ИСД – единственная терапия с доказанной эффективностью в предотвращении внезапной аритмической сердечной смерти.
Имплантация дефибриллятора необходима в случае обнаружения СБ характеристиками высокого риска. Стратификация риска основана на оценке ряда объективных клинических параметров (электрокардиографических, неинвазивных, клинических параметров, полученных в результате электрофизиологического исследования) вместе с наличием симптомов, относящихся к потенциально летальным аритмиям.
Учитывая возможное наличие наджелудочковых аритмий или нарушений, обычно использование внутриполостного дефибриллятора предпочтительнее, хотя у молодых субъектов или у пациентов, у которых возникли проблемы в связи с внутриполостным дефибриллятором, можно выбрать имплантацию подкожного дефибриллятора.
Для оптимизации наиболее подходящего терапевтического выбора на основе клинических потребностей каждого отдельного пациента всегда необходим подход, основанный на множестве параметров. Фактически, каждый из этих выборов основан на оценке риска развития потенциально летальных аритмий, которую проводят как во взрослой, так и в педиатрической популяции. В последнем контексте в самых серьезных и наиболее рискованных случаях возможна имплантация дефибриллятора. В условиях явно меньшего риска можно предложить имплантацию кардиомонитора под кожу (имплантируемый кардиомонитор ИКМ, более известный как петлевой регистратор), который постоянно записывает электрокардиограмму. Последний вариант позволяет наблюдать за пациентами на протяжении многих лет с целью документирования любых аритмий, которые могут изменить оценку аритмического риска с течением времени, и, следовательно, изменить терапевтическую стратегию, если это необходимо.
Каждое из этих устройств, ИСД или ИКМ, доступно в сочетании с технологией удаленного мониторинга, более известной как домашний мониторинг. Данная технология позволяет контролировать работу этих устройств дистанционно, пока пациент находится дома. Модем, подключенный по беспроводной связи, записывает информацию с имплантированного устройства и отправляет ее на защищенный веб-сайт, доступ к которому имеют только врачи. Таким образом, врачи могут проверить не только правильность работы устройства, но и начало любой аритмии, чтобы оперативно вмешаться, используя наиболее подходящие терапевтические стратегии.В 2015 году наша группа под руководством профессора Паппоне доказала, что аритмогенный субстрат синдрома Бругада преимущественно расположен в эпикардиальной области отводящих путей и передней стенки правого желудочка. Эти области характеризуются наличием аномальных электрических потенциалов, которые могут привести к возникновению аритмий, способных спровоцировать остановку сердца у пациентов с СБ.
После точного выявления эпикардиального аритмогенного субстрата наша команда всегда подтверждала, что эпикардиальная абляция радиочастотным методом способна устранить все электрические аномалии, что приводит к немедленному исчезновению электрокардиографической картины синдрома Бругада и отсутствию желудочковых аритмий при последующем наблюдении.
Данную процедуру предложили и разработали в нашем центре, который является самым опытным центром в мире по данному виду вмешательства.
В настоящее время в нашем центре проводится проспективное исследование для оценки эффективности и безопасности этого вида вмешательства.
Сегодня более 600 пациентов с синдромом Бругада перенесли абляцию, при этом частота периоперационных осложнений была крайне низкой.
Стратегия абляции, предложенная нашей группой может быть безопасной и эффективной у большего числа пациентов с синдромом Бругада, отнесенных к группе риска.
Этот опыт является крупнейшей в мире историей болезни пациентов с СБ, прошедших абляцию, и определяет первый шаг для новой стратегии лечения СБ, доступной для пациентов с данным состоянием.Синдром Brugada. Генетические основы синдрома Бругада
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии» Минздрава России
Синдром Бругада и синдром ранней реполяризации: различные клинические формы синдрома J-волны на примере одной семьи
Журнал: Кардиологический вестник. 2022;17(2): 81‑87
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
Наследственные каналопатии являются частой причиной внезапной смерти молодых людей. Синдром Бругада, описанный в 1992 г., — одна из разновидностей таких каналопатий. Накоплен значительный объем информации относительно эпидемиологии и патогенеза этого заболевания. Согласно консенсусу экспертов синдром Бругада вместе с синдромом ранней реполяризации объединены в единый синдром J-волны на основе общности патогенетических механизмов, однако имеется и ряд отличий. Представленный в статье случай демонстрирует различные клинические формы синдрома J-волны на примере одной семьи.
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России
ФГБУ «Национальный медицинский исследовательский центр кардиологии» Минздрава России
Дата принятия в печать:
Введение
Ежегодно в мире от сердечно-сосудистых причин умирают 17 млн человек и около 25% этих случаев приходятся на внезапную сердечную смерть (ВСС) [1]. Вероятность наступления ВСС увеличивается с возрастом. При этом случаи ВСС у молодых людей не являются большой редкостью. Согласно официальной статистике ежегодно ВСС становится причиной смерти 1100—9000 молодых людей в Европе и 800—6200 — в США [2, 3]. В 40% случаев внезапных неожиданных смертей у людей моложе 35 лет определить заболевание сердца по результатам аутопсии не удается. Основная причина ВСС в таких случаях — злокачественные желудочковые аритмии, которые могут быть обусловлены генетически детерминированными каналопатиями. Идентификация генетических мутаций в белках, ответственных за формирование и работу ионных каналов, открыло новую область трансляционных исследований в области электрофизиологии сердца [4—6].
Генетически детерминированный синдром, сопряженный со случаями ВСС, был описан в 1992 г. братьями Педро и Джозефом Бругада. Исследователями представлены 8 случаев ВСС у пациентов с элевацией сегмента ST в правых грудных отведениях и блокадой правой ножки пучка Гиса [7]. С момента описания синдрома Бругада потенциальная злокачественность его течения не вызывала сомнений. В то же время наличие изменений на электрокардиограмме (ЭКГ), характеризующихся подъемом точки J и сегмента ST в 2 или более смежных отведениях и соответствующих критериям феномена ранней реполяризации, долгое время считалось доброкачественным состоянием. Лишь к 2008 г. прогностическое значение синдрома ранней реполяризации (СРР) было пересмотрено в связи с выявленной ассоциацией этого ЭКГ-феномена с идиопатической фибрилляцией желудочков [8]. Этимм синдромам свойственно наличие мутации в генах, которое кодирует ионные каналы и приводит к нарушению фазы ранней реполяризации. Схожий патогенетический механизм формирования послужил основанием для объединения синдромов Бругада и СРР в так называемый синдром J-волны. Клинический пример сочетания синдрома Бругада и СРР приведен в нашем наблюдении.
Клинический случай
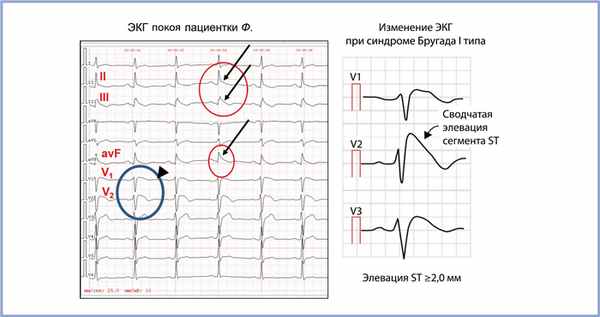
По данным ЭКГ и холтеровского мониторирования ЭКГ (ХМЭКГ) у пациентки отмечались синусовый ритм с нормальными частотными характеристиками ритма и отсутствие значимых желудочковых нарушений ритма. На ЭКГ в состоянии покоя (рис. 1), а также при ХМЭКГ (рис. 2), как и на ранее представленных ЭКГ, выявлены изменения в правых грудных отведениях в виде неполной блокады правой ножки пучка Гиса с элевацией сегмента ST по типу «свода», соответствующие синдрому Бругада I типа.
![]()
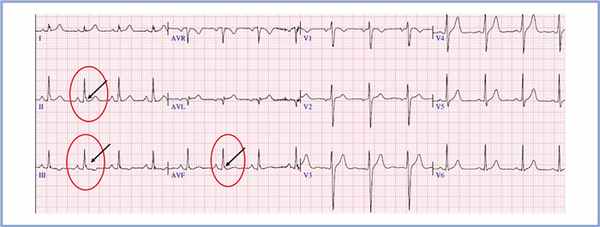
Рис. 1. ЭКГ покоя пациентки Ф.
Отмечается элевация сегмента ST в отведениях V1—V2, соответствующая синдрому Бругада I типа (указано короткой стрелкой); J-волна, преходящая элевация ST в отведениях II, III, avF (указана стрелками), соответствующая синдрому ранней реполяризации.
![]()
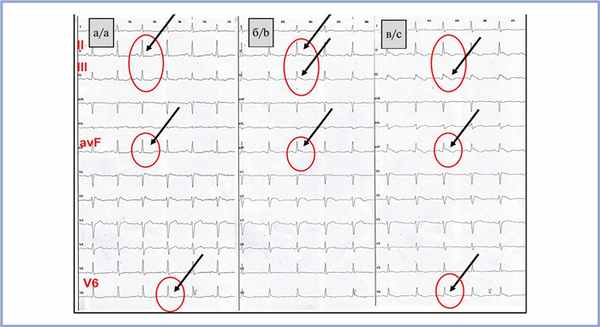
Рис. 2. Фрагменты записи холтеровского мониторирования ЭКГ в 12 отведениях пациентки Ф. в течение 24 ч. Усугубление «бругадопододобных» изменений — элевации сегмента ST по типу «свода» в отведениях V1—V2.
а — динамический характер изменений морфологии комплекса QRS в отведениях II, III, avF, V6: отсутствие признаков J-волны; б — начальные признаки J-волны; в — J-волна, косо-нисходящая элевация сегмента ST в отведениях II, III, avF, соответствующая синдрому ранней реполяризации (указана стрелками).
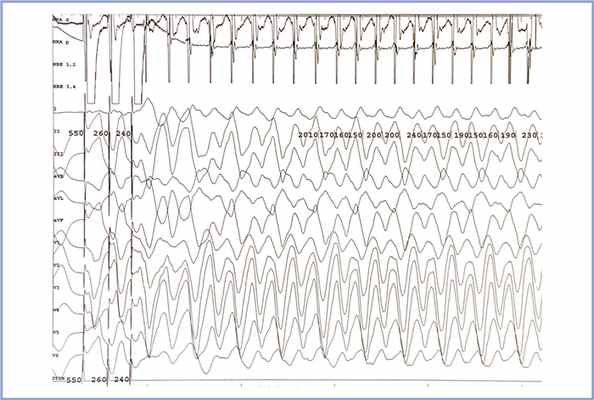
Однако наряду с этими изменениями на ЭКГ покоя (рис. 1), а также по данным ХМЭКГ (рис. 2) наблюдалось появление отчетливой J-волны и преходящей косонисходящей элевации сегмента ST в отведениях II, III, avF, V6. Элевация сегмента ST в этих отведениях не укладывалась в классическое описание синдрома Бругада. В связи с наличием изменений на ЭКГ в виде элеваций сегмента ST в правых прекордиальных отведениях для дифференциального диагноза с целью исключения патологии выполнена магнитно-резонансная томография (МРТ) с контрастированием, по результатам которой значимой структурной патологии миокарда не выявлено. Для уточнения природы пресинкопальных состояний у пациентки с семейным анамнезом ВСС проведено внутрисердечное электрофизиологическое исследование сердца. При программной стимуляции из области верхушки правого желудочка индуцированы пробежки желудочковой тахикардии (ЖТ) длительностью до 6 комплексов, при стимуляции в выносящем тракте правого желудочка — неустойчивая двунаправленная ЖТ, длительностью 15 с с частотой сокращений желудочков 290 уд/мин, сопровождающаяся головокружением, пресинкопальным состоянием и чувством нехватки воздуха, купировавшаяся самостоятельно. Этот эпизод полностью соответствовал спонтанным эпизодам «потемнения в глазах», предъявляемым в качестве жалоб (рис. 3).
![]()
Рис. 3. Эпизод индукции неустойчивой двунаправленной желудочковой тахикардии длительностью 15 с с частотой сокращений желудочков 290 уд/мин при стимуляции в выносящем тракте правого желудочка двойным экстрастимулом.
Согласно Шанхайской шкале баллов, предложенной консенсусом экспертов в 2017 г. [9], пациентке поставлен клинический диагноз: «синдром Бругада I типа. Нарушение ритма сердца: неустойчивые пароксизмы двунаправленной желудочковой тахикардии. Пресинкопальные состояния».
![]()
Рис. 4. ЭКГ второго сына пациентки Ф., 1996 года рождения.
Стрелками указана J-волна в виде зазубрины в отведениях II, III, avF (указана стрелкой), соответствующая синдрому ранней реполяризации. Изменения в правых грудных отведениях отсутствуют.
В ходе генетического исследования крови пациентки Ф., выполненного в ФГБНУ «РНЦХ им акад. Б.В. Петровского», проведен поиск генетических вариантов в последовательностях генов KCNQ1, KCNE1,KCNE2, KCNE3, KCNJ2, KCNH2, STNA1, SCN5A, SCN1B, SCN3B, SCN4B. Выявлен вероятно патогенный генетический вариант NM_198056:c.4693del (p.L1565Cfs*66) в гетерозиготном состоянии в гене SCN5A, имеющий возможное отношение к фенотипу. Аналогичная мутация обнаружена и у младшего сына пациентки, однако с учетом отсутствия каких-либо жалоб, а также признаков синдрома Бругада I типа на ЭКГ, от имплантируемого кардиовертер-дефибриллятора (ИКД) в настоящее время решено воздержаться. Проводится динамическое наблюдение. За год последующего наблюдения у пациентки по данным контрольного анализа параметров работы ИКД эпизодов устойчивой ЖТ не выявлено.
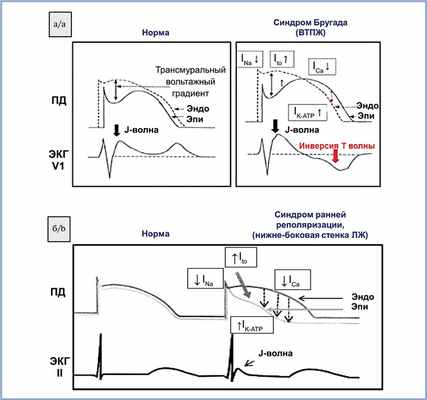
Дифференциальный диагноз синдрома Бругада требует исключения длинного ряда патологических состояний, таких как аритмогенная дисплазия правого желудочка, атипичная блокада правой ножки пучка Гиса, острый перикардит/миокардит, острая ишемия миокарда или инфаркт, тромбоэмболия легочной артерии, стенокардия Принцметала, диссекция аневризмы аорты, мышечная дистрофия Дюшенна, атаксия Фридериксена, спинотублярная мышечная атаксия, миотоническая дистрофия, механическая компрессия выносящего тракта правого желудочка (pectus excavatum, медиастинальная опухоль, гемоперикард). Большая часть этих заболеваний была исключена в ходе проведения ЭхоКГ и МРТ сердца. Гораздо более сложным является проведение дифференциального диагноза в рамках синдрома J-волны, в который помимо синдрома Бругада входит также СРР. В 2017 г. опубликован консенсус экспертов, посвященный диагностике и лечению этих редких генетических состояния, согласно которому синдромы объединяют вольтажные изменения фазы ранней реполяризации, способствующие развитию полиморфной ЖТ и фибрилляции желудочков, приводящих к ВСС у молодых людей без структурных заболеваний сердца [9]. Для синдрома Бругада характерна элевация сегмента ST в правых грудных отведениях, в то время как для СРР — элевация сегмента ST или наличие J-волны в виде зазубрины в отведениях II,III, avF, V4, V5, V6, avL, I. Механизм формирования ЭКГ-изменений является общим для этих двух синдромов. Формирование J-волны или элевации точки J служит отражением трансмурального вольтажного градиента, формирующегося за счет преобладания тока Ito в эпикардиальных слоях желудочков, однако потенциал действия в эндокардиальных слоях левого желудочка остается неизменным (рис. 5).
![]()
Рис. 5. Электрофизиологические механизмы формирования трасмурального вольтажного градиента, отражением которого является формирование J-волны или элевации точки J при синдроме Бругада в выносящем тракте правого желудочка (а) и при синдроме ранней реполяризации в нижне-боковой стенке ЛЖ (б).
Преобладание тока Ito в эпикардиальных слоях желудочков.
Существует анатомическая предрасположенность к формированию трансмурального градиента. Считается, что для СРР предположительной зоной формирования трансмурального градиента является нижняя стенка левого желудочка, однако морфологический субстрат, ответственный за формирование желудочковых аритмий, в настоящее время не обнаружен в отличие от синдрома Бругада. Так, при классическом синдроме Бругада аритмический субстрат локализован в выносящем тракте правого желудочка и представляет собой утолщение слоя эпикардиально расположенного коллагена, выраженный интерстициальный фиброз, а также ультрастуктурные изменения кардиомиоцитов. Для синдрома Бругада характерно значительное уменьшение экспрессии коннексина 43, в норме способствующего электрическому сопряжению каридомиоцитов. Электрическое разобщение миоцитов из-за уменьшения экспрессии коннексина 43 на вставочных дисках и тонкого интерстициального и замещающего фиброза приводит к задержке проведения в выносящем тракте правого желудочка. В результате ЭКГ эпикарда становятся ненормальными, замедленными и фрагментированными. Это обеспечивает основу для формирования ЭКГ-картины Бругада типа I и генерации полиморфной желудочковой тахикардии и фибрилляции желудочков [10]. Наличие четкого анатомического субстрата у синдрома Бругада делает возможным и эффективным применение абляции у пациентов с повторными разрядами ИКД.
Проводя параллели между основными сходствами и отличиями синдромов Бругада и синдромов ранней реполяризации, необходимо отметить, что для обоих синдромов характерно развитие клинических проявлений чаще у мужчин, начиная с третьей декады жизни. Также свойственны высокая изменчивость ЭКГ-проявлений, возникновение фибрилляции желудочков во сне или при низкой физической активности, способствующей появлению ЖЭС с коротким интервалом сцепления, высокая чувствительность желудочковых нарушений ритма к хинидину. Основными отличиями считаются разные ЭКГ-отведения, в которых эти два синдрома демонстрируют свои проявления, различная реакция ЭКГ-изменений на применение блокаторов натриевых каналов (манифестация J-волны в случае синдрома Бругада, исчезновение J-волны при СРР), а также основные генетические мутации, приводящие к развитию синдромов. Мутации в генах, кодирующих белки натриевых каналов SCN5A (как в описании случая пациентки Ф.), SCN10A, SCN3В, являются наиболее распространенными среди пациентов с синдромом Бругада и обнаруживаются у 11—28%, 5—16,7% и 1,1% пробандов соответственно. Однако в ряде случаев при синдроме Бругада находят и другие генетические мутации, кодирующие белки кальциевых каналов и аналогичные мутациям, характерным СРР. Так, мутации в генах CACNA1C, CACNB2b, CACNA2D1 находят в 6,6; 4,8 и 1,8% при синдроме Бругада и в 4,1; 8,3 и 4,1% при СРР. Их наличие в дополнение к мутациям белков натриевых каналов, вероятно, может иметь аддитивный эффект и усугублять проявления синдрома J-волны [9].
Наиболее часто в популяции встречаются пациенты с признаками J-волны на ЭКГ в боковых или нижнебоковых отведениях. Считается, что риск развития фатальных аритмических событий таких пациентов невысок. При присоединении к изменениям в боковых и нижнебоковых отведениях «бругадоподобных» изменений ЭКГ — подъема сегмента ST по типу «свода» в правых прекордиальных отведениях — риск внезапной смерти пациента возрастает. При этом достигает своего максимума у пациентов с глобальными изменениями ЭКГ при сочетании «бругадоподобных» изменений с наличием высокоамплитудной J-волны в нижних и боковых отведениях. К данной крайне высокой группе риска относится пациентка Ф. Ее единственный оставшийся в живых сын входит в группу с умеренным риском аритмических событий, но требует тщательного динамического наблюдения. Это обусловлено тем, что отдельными исследователями продемонстрирована возможность последовательного усугубления ЭКГ-проявлений синдрома J-волны в течение жизни [11].
Представленное клиническое наблюдение демонстрирует разнообразие клинических форм и ЭКГ-проявлений синдрома J-волны, а также тесную взаимосвязь считавшегося долгое время доброкачественным СРР и потенциально злокачественного синдрома Бругада. В таких случаях решение вопроса о необходимости применения мер профилактики ВСС должно приниматься на основании индивидуальной оценки степени риска и потенциально может быть пересмотрено в течение жизни пациента. Своевременный пересмотр стратегий лечения подобных пациентов может быть осуществлен только при динамическом наблюдении. Фенотипические проявления одних и тех же генетических мутаций при разных вариантах синдрома J-волны могут иметь различия даже у членов одной семьи, варьируя от случаев внезапной смерти до малосимптомных и бессимптомных форм.
Синдром Бругада
Синдром Бругада – наследственное заболевание, которое приводит к аритмии, но характеризуется отсутствием структурной патологией сердца. При этой патологии появляются признаки блокады правой ножки пучка Гиса и могут возникать жизнеугрожающие желудочковые аритмии. Эти состояния повышают риск внезапной сердечной смерти.
Заболевание впервые описано в 1992 году испанскими учеными – братьями Педро и Джозефом Бругада. Они описали ЭКГ-картину трехлетней девочки в сочетании с семейным анамнезом и клинической картиной заболевания. Ребенок страдал от обмороков. В семейном анамнезе был случай внезапной сердечной смерти – от остановки сердца умер родной брат девочки.
Причины синдрома Бругада
Причины болезни – мутации генов. Первый мутировавший ген установили в 1998 году. С тех пор врачи уже нашли более 450 возможных изменений в 24 генах, которые ассоциированы с синдромом Бругада.
Хотя список генов постоянно расширяется, только у одного из трех пациентов с этим заболеванием возможно генетическое подтверждение диагноза. В остальных случаях причины тоже наследственные. Проблема генетической диагностики в том, что установлены не все гены, отвечающие за развитие болезни.
Аритмия связана нарушением функции трансмембранных ионных каналов. Через них проходят электролиты, обеспечивающие процессы возбуждения в миокарде. Таких веществ четыре: натрий, калий, кальций и магний. Чаще всего страдает функция доставки натрия в кардиомиоциты (клетки сердца). В итоге мышца не может полноценно расслабиться. Сильнее от такого нарушения страдает правый желудочек.
Истинная распространенность наследственного синдрома неизвестна. Большинство случаев попросту не диагностируют в течение жизни. Распространенность заболевания в Европе оценивается разными авторами от 1 до 5 случаев на 10 тысяч человек. Болезнь значительно чаще встречается среди населения Кавказа и бурятов. Синдромом Бругада не страдают люди африканского происхождения.
Мужчины болеют в 3 раза чаще женщин. Объясняется это тем, что у них болезнь чаще вызывает симптомы и осложнения. В то же время у женщин она чаще протекает бессимптомно из-за особенностей гормонального профиля.
Классификация синдрома Бругада
Болезнь классифицируют по выявляемым с помощью ЭКГ изменениям. Две формы синдрома:
В свою очередь неполная форма может быть четырех типов:
- типичная ЭКГ-картина синдрома Бругада, но нет отягощенного семейного анамнеза и отсутствуют симптомы;
- изменения на ЭКГ, в семейном анамнезе нет указаний на полную форму синдрома;
- типичные изменения на ЭКГ после фармакологических тестов;
- наличие у больного синкопе (обмороков) и фибрилляции предсердий в анамнезе.
Клиническое течение заболевания бывает:
- синкопальное (с обмороками);
- бессинкопальное (без обмороков).
По результатам генетической диагностики синдром Бругада бывает:
- генотип-позитивный;
- генотип-негативный.
Симптомы синдрома Бругада
Вылечить болезнь невозможно.
Задача врачей состоит в том, чтобы оценить риск внезапной сердечной смерти. Если он высокий, то пациенту имплантируют кардиовертер-дефибриллятор. Это устройство, которое отслеживает сердечный ритм. В случае возникновения жизнеугрожающей желудочковой тахикардии оно подает разряд, который восстанавливает нормальный сердечный ритм.
Максимальный риск отмечается у пациентов, выживших после остановки сердца. Это первые кандидаты на имплантацию кардиовертера-дефибриллятора.
К группе промежуточного риска относятся пациенты с синкопальным течением заболевания (обмороками).
Медикаментозная терапия применяется только в случае электрического шторма. Это ситуации, когда желудочковые аритмии возникают слишком часто, из-за чего кардиовертер-дефибриллятор дает постоянные разряды. В итоге ухудшается качество жизни пациента. К тому же, аппарат разряжается быстрее.
Используются препараты, которые подавляют калиевые каналы, усиливают натриевые и кальциевые ионные каналы. Основные препараты: хинидин и изопротеренолол. Но доказательства эффективности этих средств ограничены.
Другие препараты, влияющие на функцию ионных каналов:
- новокаинамид;
- аймалин;
- флекаинид;
- пропафенон.
Однозначно нет смысла использовать бета-адреноблокаторы и амиодарон. По результатам исследований сделан однозначный вывод о неэффективности этих средств.
Эпикардиальная катетерная абляция – новый вариант лечения пациентов с синдромом Бругада. В Европе эта процедура проводится с использованием системы системы трехмерного навигационного картирования. Операцию делают тем, у кого ранее установленный кардиовертер-дефибриллятор дает частые разряды.
Суть процедуры состоит в разрушении небольшого участка сердца, который становится источником патологических импульсов. Абляция бывает эпикардиальная и эндокардиальная. Обычно в медицине применяется эндокардиальная абляция – она проводится изнутри сердца, а доступ к нему получают через кровеносные сосуды. Но при синдроме Бругада требуется эпикардиальная абляция – разрушение очага снаружи сердца. Для этого требуется разрез на груди или в верхней части живота.
В Европе эпикардиальная катетерная абляция выполняется через субксифоидальный доступ.
В отличие от традиционного разреза между ребрами, при таком подходе разрез делают под грудиной. В результате нет риска повреждения ребра, надкостницы, межреберного нерва, а это основная причина послеоперационной боли.
Прогноз синдрома Бругада
Большинство людей с синдромом Бругада никогда не страдают от аритмий. Риск внезапной сердечной смерти очень низкий.
При первом описании синдрома было установлено, что вероятность внезапной сердечной смерти или фибрилляции желудочков в течение 3 лет после установления диагноза составляет 32%. Но такая статистика связана с дефицитом диагностики. Выявлялись только наиболее явные случаи заболевания. Сегодня врачи умеют диагностировать даже бессимптомные формы. В 2 случаях из 3 синдром Бругада выявляется у пациентов, которые не имеют в анамнезе желудочковых аритмий и обмороков.
Наличие синкопе повышает риск фибрилляции предсердий и внезапной сердечной смерти в 4 раза, а остановка сердца в анамнезе – в 16 раз по сравнению с бессинкопальным течением заболевания.
Профилактика синдрома Бругада
Профилактика невозможна. Болезнь передается по наследству.
Для предотвращения жизнеугрожающих желудочковых аритмий стоит по возможности не использовать лекарственные средства, нарушающие функцию ионных каналов.
Не стоит употреблять наркотики, особенно кокаин, и злоупотреблять алкоголем.
Пациентам с синдромом Бругада врачи часто рекомендуют отказаться от соревновательного спорта, связанного с экстремально высокими физическими нагрузками. Считается, что интенсивная нагрузка или восстановление после неё связано с высоким риском возникновения желудочковой аритмии. Пока что вред профессионального спорта доказан только для симптомных пациентов. Нет убедительных доказательств, что высокие нагрузки могут навредить людям, у которых заболевание никогда не проявлялось обмороками или другими симптомами.
При наличии синдрома Бругада, необходима индивидуальная оценка риска тяжелых желудочковых аритмий. Если он высокий, то профилактика внезапной сердечной смерти осуществляется путем имплантации кардиовертера-дефибриллятора.
Статья подготовлена по материалам:
Синдром Бругада, клинические рекомендации Ассоциации сердечно-сосудистых хирургов России, 2020
HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes, 2013
Antzelevitch C., Yan G.X. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm, 2015
Curcio A., Mazzanti A., Bloise R., Monteforte N., Indolfi C., Priori S.G., Napolitano C. Clinical presentation and outcome of Brugada syndrome diagnosed with the new 2013 criteria. J. Cardiovasc. Electrophysiol, 2016
Читайте также: