Синдром Чедиака-Хигаси
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Тип наследования — аутосомно-рецессивный. Относится к группе наследственных нарушений функции фагоцитирующих клеток. Клинически может быть заподозрен в первые месяцы и годы жизни при наличии клинической картины, также рецидивов неясной лихорадки, частых инфекций дыхательных путей, желудочно-кишечного тракта, кожи.
Клинические проявления разнообразны: рецидивирующие инфекции (ОРЗ, бронхиты, пневмония, отит, синуситы, абсцессы). Обычно инфекционные осложнения вызываются микробной флорой (стафилококк, Гр-), реже грибковый. Приблизительно у 1/3 больных выявляются геморрагии, повышение температуры тела при отсутствии инфекции.
У больных отмечается частичный альбинизм волос, кожи, окраски глаз. У них светлая прозрачная кожа с тонкими сухими светлыми волосами пепельного, серебристого или свинцового цвета. Радужная оболочка светлая, на сетчатке — пигментация, отмечается нистагм. Характерен универсальный гипергидроз и светобоязнь.
Если заболевание протекает длительно, то характерны нарушения ЦНС: парезы, нарушения чувствительности, гипо- и арефлексия, мозжечковые нарушения, УО.
У большинства детей до 10 лет возникает острая (торпидная) фаза: повышение температуры, аденопатия, гепатоспленомегалия, геморрагический синдром, — связаны с тромбоцитопенией и нарушением функции нейтрофилов. Большинство детей в эту фазу умирают от геморрагического синдрома или сепсиса. Иначе эту стадию называют стадией акселерации. Она может наступить в любом возрасте, от новорожденности до пубертата.
Морфологически характеризуется лимфогистиоцитарной инфильтрацией печени селезенки, лимфоузлов, тимуса с явлениями гемофагоцитоза различной выраженности. Необходимо отметить, что при этом синдроме обязательно исследуют спинномозговую жидкость, где также обнаруживается эритрофагия.
Характерная особенность синдрома — наличие гигантских пероксидазоположительных гранул в нейтрофилах, эозинофилах, моноцитах периферической крови и костном мозге, в клетках предшественницах гранулоцитов, которые содержат дегенеративные вакуоли. Гранулы появляются в результате слияния первичных и вторичных лизосом. Несмотря на высокий уровень в них пероксидазы, нарушение слияния с фагосомами препятствует завершению фагоцитоза, так как гигантские лизосомы не способны передавать свои гидролитические ферменты в фагосомы нейтрофилов, содержащих неинтегрированные бактерии. Это предрасполагает к бактериальным инфекциям. При этом заболевании фагоцитарная активность нейтрофилов и меланоцитов нормальные, а хемотаксис и переваривающая способность снижены. Это может привести к тому, что нейтрофилы могут стать «убежищем» для бактерий от антибиотиков и других фагоцитирующих клеток.
Патогенез синдрома связан с наличием аномалии мембраны клеток. Поэтому возникает неконтролируемое слияние лизосом, нарушение хемотаксиса нейтрофилов, изменение функции тромбоцитов, снижение естественной киллерной активности лимфоцитов, снижение АЗКЦ. Большая часть клинических проявлений объясняется ненормальным распределением лизососмальных ферментов. Повреждение клетки характеризуется изменением структурно-химических свойств, метаболизма, структуры и функций клетки, которые ведут к нарушению к ее жизнедеятельности.
Клетка является открытой саморегулирующейся системой. Структура нормальной клетки направлена на осуществление определенного метаболизма, дифференцировку и специализацию. Различные патогенные агенты при воздействии на клетку могут вызвать следующие процессы: адаптацию, повреждение, гибель.
Виды повреждения
4. Необратимое (смерть). Существует 2 типа клеточной смерти: некроз и апоптоз.
Причины повреждения
1. Физические (колебания температуры, механическая травма, ионизирующая радиация, электрический шок).
2. Химические (яды, лекарственные вещества, факторы окружающей среды).
3. Биологические (инфекционные агенты, иммунные реакции, генетические нарушения, дисбаланс питания).
По происхождению:
1. Экзогенные и эндогенные.
2. Инфекционные и неинфекционные.
Действие повреждающих факторов может быть прямым и опосредованным.
Прямое повреждающее воздействие оказывают следующие факторы: яды (цианистый калий), аноксия, очень низкие значения рН, недостаток ионов кальция, ионизирующая радиация. При опосредованном повреждении развиваются вторичные реакции, образуются медиаторы повреждения либо другие вещества-посредники. Часто первичные изменения при повреждении остаются неизвестными. Характер ответа клетки на повреждение зависит от ее гормонального статуса, характера питания и метаболических потребностей. Реакция клетки на повреждающий агент зависит от типа, продолжительности действия и степени тяжести повреждающего агента (например, глюкоза и поваренная соль в повышенных концентрациях, способны вызвать повреждения клеток путем нарушения электролитного гомеостаза).
Патология клетки является интегративным понятием, включающим патологию клеточных ультраструктур, и компонентов, механизмы структурно-функциональных нарушений жизнедеятельности клетки, нарушение межклеточных взаимодействий и кооперации клеток при общепатологических процессах.
Механизмы повреждения клетки
1. Повреждение мембранного аппарата и ферментных систем клетки.
2. Дисбаланс ионов и жидкости в клетки.
3. Нарушение энергетического обеспечения клеточных процессов.
4. Нарушение генетической программы клетки и механизмов ее реализации.
5. Расстройства внутриклеточных механизмов регуляции функции клетки.
Патология клеточного ядра
К патологии клеточного ядра относятся следующие состояния:
1. Патология самого ядра (изменения размеров и структуры ядра, формы, количества ядер и ядрышек, появление ядерных включений).
2. Патология ядерной мембраны.
3. Патология митоза.
Изменения структуры ядра
Полиплоидия — увеличение числа хромосом до величины, кратной их нормальному гаплоидному набору (23 хромосомы). Таким образом, при триплоидии общее число хромосом равно 69, при тетраплоидии — 92 и т.д. При полиплоидии процесс репродукции не достигает типичного эндоцитоза. Полиплоидия развивается при редупликации ДНК и отсутствии спирализации хромосом.
Полиплоидные клетки выявляются:
1. В нормально функционирующих органах, тканях и клетках человека: в печени, почках, миокарде, эпидермисе мегакариоцитах, гигантских клетках трофобласта.
2. При старении организма.
3. При репаративной регенерации (печень), при компенсаторной гипертрофии (миокард).
4. При опухолевом росте.
Способы выявления полиплоидии:
1. По размеру ядра.
2. По увеличению количества ДНК в интерфазном ядре.
3. По увеличению числа хромосом в митатической клетке.
Анеуплодия — изменение в виде неполного набора хромосом.
При анеуплодии происходит рост или снижение общего числа хромосом в генотипе организма по отношению к его нормальной величины. При этом изменения не захватывают каждую хромосому в нормальном гаплоидном наборе. Анеуплодия связана с хромосомными мутациями. При анеуплодии может меняться число аутосом и количество половых хромосом. Проявление анеуплодии обнаруживаются злокачественных опухолях.
Микротельца (пероксисомы)
Пероксисомы являются вспомогательной системой окисления в клетке. Изменения микротелец отражают нарушения оксидазно-каталазной активности клеток. При повреждении клетки могут наблюдаться следующие изменения микротелец:
1. Первичные — «пероксисомные болезни».
2. Вторичные — изменение числа и структурных компонентов пероксисом.
Пероксисомные болезни
1. Акаталаземия. Характеризуется резким снижением активности каталазы в печени и других органах. Клинически проявляется изъязвлениями в полости рта.
2. Цереброгепаторенальный синдром Целлвегера. Характеризуется отсутствием пероксисом в гепатоцитах. Нарушен синтез желчных кислот.
3. Системная недостаточность карнитина. Характеризуется выраженным дефицитом карнитина в различных органах и тканях. Клинически проявляется миопатией, нарушением функций печени и головного мозга.
Увеличение числа пероксисом возникает при алкогольной интоксикации. Снижение числа пероксисом наблюдается при гипоксии, воздействие ионизирующего излучения. Разрушение пероксисомного матрикса происходит при перевязки печеночных вен, вирусном гепатите, ишемическом некрозе, гиперлипидемии, гиперхостеринемии, при опухолевом росте.
Синдром Чедиака-Хигаси (обзор литературы и собственные клинические наблюдения)
Синдром Чедиака-Хигаси (СЧХ) - редкое иммунодефицитное состояние с аутосомно-рецессивным типом наследования, обусловленное мутацией в гене LYST/CHS1, кодирующем соответствующий белок-регулятор лизосомального транспорта. СЧХ характеризуется ранним дебютом, яркой клинической картиной и определенными лабораторными признаками (глазо-кожный альбинизм, гигантские пероксидазаположительные гранулы в гранулосодержащих клетках) и высоким риском развития гемофагоцитарного лимфогистиоцитоза (фазы «акселерации»), в большинстве случаев являющегося фатальным. Трансплантация гемопоэтических стволовых клеток (ТГСК) - единственный метод излечения данного заболевания. Лучшие результаты общей выживаемости пациентов с СЧХ достигнуты при выполнении ТГСК до развития фазы «акселерации», а также с использованием режимов кондиционирования со сниженной интенсивностью. В статье представлен опыт диагностики и лечения трех пациентов с СЧХ с различной степенью тяжести и осложнениями основного заболевания. Согласно международным рекомендациям двум пациентам выполнена ТГСК с режимом кондиционирования со сниженной интенсивностью. По результатам катамнеза (длительность наблюдения 4 мес и 1 год) достигнута полная гематологическая реконституция.
Ключевые слова
Об авторах
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России
Россия
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России
Россия
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России
Россия
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России
Россия
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России
Россия
Список литературы
1. Islam AS, Hawsawi ZM, Islam MS, Ibrahim OA. Chédiak-Higashi syndrome: an accelerated phase with hereditary elliptocytosis: case report and review of the literature. Ann Saudi Med. 2001; 21(3-4): 221-4.
2. Beguez-Cesar AB. Neutropenia crönica maligna familiar congranulaciones atlpicas de los leucocitos. Boletln de la Sociedad Cubana dePediatrla. 1943; 15: 900-22.
3. Chediak MM. New leukocyte anomaly of constitutional and familial character. Rev Hematol. 1952; 7(3): 362-7.
4. Higashi O. Congenital gigantism of peroxidase granules: the first case ever reported of qualitative abnormity of peroxidase. Tohoku J Exp Med. 1954; 59(3): 315-32.
5. Sato A. Chédiak and Higashi's disease: probable identity of a new leucocytal anomaly (Chédiak) and congenital gigantism of peroxidase granules (Higashi). Tohoku J Exp Med. 1955; 61(2-3): 201-10.
6. Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996; 382(6588): 262-5.
7. Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008; 15(1): 22-9.
9. Ochs HD, Smith CIE, Puck JM, eds. Primary immunodeficiency diseases: a molecular and cellular approach. 3rd ed. USA, New York: Oxford University Press: 2014: 749-50.
10. Huynh C, Roth D, Ward DM, Kaplan J, Andrews NW. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci USA. 2004; 101(48): 16795-800.
12. Lozano ML, Rivera J, Sanchez-Guiu I, Vicente V. Towards the targeted management of Chediak-Higashi syndrome. Orphanet J Rare Dis. 2014; 9: 132.
13. Nargund AR, Madhumathi DS, Premalatha CS, Rao CR, Appaji L, Lakshmidevi V. Accelerated phase of Chediak Higashi syndrome mimicking lymphoma-- a case report. J Pediatr Hematol Oncol. 2010; 32(6): e223-6.
15. Dotta L, Parolini S, Prandini A, Tabellini G, Antolini M, Kingsmore SF, et al. Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculocutaneous albinism. Orphanet J Rare Dis. 2013; 8: 168.
16. Mehta RS, Smith RE. Hemophagocytic lymphohistiocytosis (HLH): a review of literature. Med Oncol. 2013; 30(4): 740.
17. Sanchez-Guiu I, Antön AI, Garcla-Barberâ N, Navarro-Fernandez J, Martinez C, Fuster JL, et al. Chediak-Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur J Haematol. 2014; 92(1): 49-58.
18. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012; 119(12): 2754-63.
19. Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010; 54(4): 563-72.
20. Jessen B, Bode SF, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type Blood. 2013; 121(15): 2943-51.
21. Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013; 160(3): 275-87.
22. Gr0nskov K, Brandum-Nielsen K, Lorenz B, Preising MN. Clinical utility gene card for: oculocutaneous albinism. Eur J Hum Genet. 2014; 22(8). doi: 10.1038/ejhg. 2013.307: published online 12 February 2014.
23. Chatzinasiou F, Stratigos A, Rigopoulos D. Pigment genodermatoses affecting melanocyte development and migration from the neural crest: piebaldism, Waardenburg syndrome and Cross-McKusick-Breen syndrome. J. Pigment. Disord. 2015; 2(3): 168. doi: 10.4172/2376-0427.1000168.
24. Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant. 2007; 39(7): 411-5.
25. Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005; 129(5): 622-30.
26. Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010; 116(26): 5824-31.
27. Marsh RA, Jordan MB, Filipovich AH. Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: an important step forward. Br J Haematol. 2011; 154(5): 556-63.
28. Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood. 2005; 106(1): 40-2.
30. Ogimi C, Tanaka R, Arai T, Kikuchi A, Hanada R, Oh-Ishi T. Rituximab and cyclosporine therapy for accelerated phase Chediak-Higashi syndrome. Pediatr Blood Cancer. 2011; 57(4): 677-80.
31. Grada A, Weinbrecht K. Next-generation sequencing: methodology and application. J Invest Dermatol. 2013; 133(8): e11.
Синдром Чедиака-Хигаси
В статье приводятся данные клинического наблюдения за пациентом с редким наследственным заболеванием – синдромом Чедиака – Хигаси. Данный синдром наследуется по аутосомно-рецессивному типу, основными признаками которого являются: рецидивирующие инфекции, частичный альбинизм, фотофобия, нистагм и наличие цитоплазматических гранул в клетках лейкоцитарного ряда. Заболевание обычно проявляется у детей младенческого возраста, которое может быстро прогрессировать, или иметь нетяжелое, рецидивирующее течение, но у детей старшего возраста переходит в ускоренную фазу с летальным исходом обычно до достижения ими 10-летнего возраста, причиной которого становятся инфекции или злокачественные новообразования. В работе показаны особенности течения заболевания у данного пациента. Установлено, что применение комплекса лечебных мероприятий позволило пациенту достигнуть 19-летнего возраста. В настоящее время у больного регистрируется прогрессирование заболевания и множественные осложнения, что предполагает дальнейший неблагоприятный прогноз.
1. Гундорова Л.В. Синдром Чедиака – Хигаси с гемофагоцитарным синдромом / Л.В. Гундорова, В.П. Нажимов, А.А. Масчан // Вестник Российского университета дружбы народов, серия: Медицина, 2000. – № 2. – С. 38–41.
2. Каракина М.Л. Анализ генеалогических данных у взрослых с первичным иммунодефицитом / М.Л. Каракина, В.Н. Шершнев, И.А. Тузанкина // Пульмонология, 2015. – № 2. – С. 2003–2010.
3. Масчан М.А., Новичкова Г.А. Гемофагоцитарный лимфогистиоцитоз // Вопросы современной педиатрии. – М., 2009. – Т. 8. – № 3. – С. 66–75.
4. Новичкова Г.А., Минков М., Масчан М.А., Чернов В.М. Гистиоцитозы // Клиническая онкогематология / под ред. М.А. Волковой. – М., 2007.
5. Тузанкина И.В., Каракина М.Л., Власова Е.В. Анализ клинических проявлений дебюта первичного иммунодефицита у взрослых // Медицинская иммунология, 2014. – № 4. – С. 364–374.
7. Henter J.I., Horne A., Arico M. et al. HLH72004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis // Pediatr. Blood Cancer. – 2007. – V. 48, № 2. – P. 124–131.
Синдром Чедиака – Хигаси (заболевание названо по имени японского врача Хигаси и кубинского Чедиак) – редкий синдром с генерализованной клеточной дисфункцией с аутосомно-рецессивным типом наследования, который относится к особой группе иммунодефицитов. Страдают в основном дети раннего возраста, смерть, причиной которой становятся инфекции или злокачественные новообразования, часто наступает до достижения ими возраста 10 лет. Заболевание клинически характеризуется тяжелыми гнойными инфекциями, альбинизмом, косоглазием, светобоязнью, нистагмом, прогрессирующей нейропатией, задержкой умственного развития, склонностью к кровотечениям. У большинства больных в течение болезни на фоне бактериальной или вирусной инфекции развивается гемофагоцитарный синдром, который клинически проявляется лихорадкой, отеками, гепатоспленомегалией, желтухой, лимфоаденопатией, панцитопенией, судорогами, комой, коагулопатией [1]. Характерным признаком данного синдрома является наличие гигантских пероксидазоположительных гранул в нейтрофилах, эозинофилах, моноцитах периферической крови и костного мозга, циркулирующих лимфоцитах, цитоплазме нейронов и клетках соединительной ткани периневральной области. Нейтрофилы больных способны нормально фагоцитировать и продуцировать супероксид-анион, но не могут осуществлять внутриклеточное переваривание фагоцитированных микроорганизмов, что проявляется тяжелыми инфекциями бактериальной или грибковой природы. Лимфоциты больных вне гемофагоцитарного синдрома обычно не отличаются от нормальных в реакциях бласттрансформации с митогенами, но контролирующая функция этих клеток почти всегда значительно нарушена [2]. Вышеуказанные нарушения, вероятно, лежат в основе развития гемофагоцитарного синдрома, при котором нарушается контроль над пролиферацией и функцией активированных лимфоцитов и макрофагов. Морфологически проявляется массивной лимфогистиоцитарной инфильтрацией многих органов и тканей с явлениями гемофагоцитоза.
Цель исследования: изучить особенности течения редкого наследственного синдрома Чедиака – Хигаси и оценить влияние комплекса проводимых лечебных мероприятий на продолжительность и качество жизни пациента.
Материалы и методы. Исследование основано на данных клинического наблюдения за пациентом, получающим лечение в отделении гематологии Республиканской клинической больницы им. Н.А. Семашко (г. Улан-Удэ).
Результаты и их обсуждение. Больной У., 19 лет диагноз: Агранулоцитоз. Первичный иммунодефицит. Синдром Чедиака – Хигаси. Афтозный стоматит. Хронический тонзиллит. Синдром Крампи. Нейропатия. Глазной альбинизм. Миопатический астигматизм. Косоглазие содружественное расходящееся. Анемия средней степени тяжести. Тромбоцитопения II степени тяжести, геморрагический синдром. Анасарка. Состояние после торакотомии слева, ревизии левой плевральной полости, перикардотомии, дренирования полости перикарда и плевры от 14.04.2016 года.
С ноября 2015 года терапия колониестимулирующими препаратами была прервана из-за их отсутствия. С марта 2016 года отмечалось резкое ухудшение состояния больного, вероятно спровоцированное перерывом в приеме колониестимулирующего препарата, на фоне чего отмечается развитие гемофагоцитарного синдрома, что подтверждается клиническими и лабораторными данными [3]. В связи с тяжестью состояния больной в экстренном порядке был госпитализирован в гематологическое отделение Республиканской клинической больницы им. Н.А. Семашко. При поступлении отмечалась выраженная слабость, одышка смешанного характера в покое, усиливающаяся при незначительной двигательной активности, утомляемость, геморрагическая сыпь по телу, на конечностях, светобоязнь, боли в костях, вздутие и боли в животе, увеличение в объеме живота, отеки нижних конечностей, отсутствие аппетита, выраженная мышечная слабость – больной не может себя обслуживать, повышение температуры тела до 38 ºС. При осмотре отмечено – состояние по заболеванию тяжелое. Сознание ясное, положение вынужденное – лежит в постели, контакту доступен, критичность снижена. Правильного телосложения, астенического типа, кахексия. Рост – 177 см, вес – 54 кг, ИМТ – 17. Кожные покровы бледные, с желтушным оттенком, обычной влажности, обильная мелкоточечная геморрагическая сыпь на коже верхних, нижних конечностей, туловище, на передней брюшной стенке гематома диаметром 4,0 см., фиолетового цвета. В крестцовой области и в области тазобедренных суставов имеются пролежни в диаметре до 3 см. Слизистые оболочки бледные, умеренной влажности, склеры иктеричные, отмечается субъконъюнктивальное кровоизлияние. Периферические лимфоузлы не пальпируются. Щитовидная железа не увеличена, безболезненна. Мышечная система и подкожно-жировая клетчатка выражены слабо. Килевидная деформация грудной клетки. Обе половины грудной клетки одинаково участвуют в акте дыхания. Нижние границы легких смещены вверх на 2 см. При перкуссии грудной клетки легочный звук, в нижних отделах притупленный. Дыхание везикулярное, в нижних отделах ослабленное, хрипов нет. ЧДД 22–24 в минуту. Визуально область сердца не изменена, верхушечный толчок не визуализируется, при пальпации расположен по левой срединноключичной линии. Границы относительной тупости сердца расширены влево: правая – IV м/р у правого края грудины, верхняя – III ребро по левой окологрудинной линии, левая – V м/р по левой срединноключичной линии. Границы абсолютной тупости сердца: правая – IV м/р у левого края грудины, верхняя – IV м/р по левой окологрудинной линии, левая – V м/р по левой срединноключичной линии. Тоны сердца ритмичные, слегка приглушены, ЧСС – 100 уд. в мин, PS – 100 в мин, АД – 130/90 мм рт. ст. Полость рта: слизистые гиперемированы. Миндалины увеличены до 1 ст., гиперемированы. Задняя стенка глотки гиперемирована, налета нет. Язык влажный, на кончике языка имеется афтозная язва размерами 0,3 х 0,4 мм. Живот увеличен в размерах за счет асцита, гепатоспленомегалии, при пальпации умеренная болезненность по всему животу. Печень +3см от края реберной дуги, при пальпации – болезненность. Селезенка пальпируется + 5 см из-под края реберной дуги, умеренная болезненность при пальпации. Стул регулярный, кашицеобразный, без патологических примесей. Мочеиспускание свободное, безболезненное. Моча светлая, суточный диурез считают, по объему достаточный. Выраженные отеки стоп и до нижней трети голеней. В дальнейшем наблюдалось ухудшение состояния больного в виде прогрессирования анемического, отечно-асцитического, гемофагоцитарного синдрома, развития полисерозита. При пункции плевральной полости и перикарда эвакуировано значительное количество жидкости с примесью крови до 1200–1500 мл. В связи с ухудшением состояния с 12.04. по 19.04. 2016 г. пациент находился в отделении интенсивной терапии. По поводу состояния больного неоднократно проводились врачебные консилиумы. Пациент продолжал получать лечение антибактериальными, противогрибковыми средствами, инфузионную терапию, препараты иммуноглобулина – пентаглобин 50 мг в/в, нутритивную и респираторную поддержку, трансфузии донорской крови и ее компонентов, мочегонные средства. 14.04.2016 г. пациенту с письменного согласия родителей было произведено оперативное вмешательство: боковая торакотомия слева с ревизией левой плевральной полости, перикардотомия, установлены дренажи в полости плевры и перикарда. Ежедневно по дренажам отделялось примерно 1000–1200 мл жидкости с примесью крови, в дальнейшем серозного характера. Оперативное вмешательство больной перенес удовлетворительно, в послеоперационный период болевой синдром умеренный, анемия не нарастала, температура периодически фебрильная. 19.04.16 г. в связи со стабильным состоянием и некоторой положительной динамикой больной был переведен в отделение гематологии. С 20 апреля появилась возможность получать колониестимулирующий препарат лейкостим в дозе 2,0 мл подкожно 1 раз в день. На фоне приема данного препарата отмечалась незначительная эскалация лейкоцитов до 1,3 х 10 9 . В дальнейшем, на фоне проводимой комплексной терапии, наметилась положительная динамика в состоянии больного в виде купирования геморрагического, анемического, отечно-асцитического синдромов. Субъективно у больного несколько улучшилось общее состояние, прошла одышка, появился аппетит, больной стал активен, стал присаживаться, несколько уменьшились симптомы интоксикации, уменьшилась лихорадка. Дренажи были удалены вследствие уменьшения выпота в полостях, а также из-за высокого риска присоединения вторичной инфекции и развития гнойных осложнений и решения консервативного ведения больного. 16.05.2016 года по просьбе пациент выписан домой с рекомендациями продолжения терапии амбулаторно и повторной госпитализации в отделение гематологии через одну неделю.
Данные результатов обследований при поступлении:
В клиническом анализе крови цитопения периферической крови, с вовлечением 2–3-х ростков кроветворения анемия – уровень гемоглобина от 55 г/л, тромбоцитопения 111х10 9 , лейкопения – от 0,52 х10 9 , СОЭ до 55 мм/ч.
В коагулограмме выявлена изолированная гипофибриногенемия, гипокоагуляция. В биохимическом анализе крови обращает внимание гипоальбуминемия до 35 г/л, гипертриглицеридемия, гипонатриемия, повышение уровня АСТ до 90, АЛТ до 50, билирубина до 107 мкмоль/л, показатели ЛДГ в пределах нормы – до 354 ед/л, повышен уровень щелочной фосфатазы до 687 ед/л, уровень креатинина в пределах нормальных величин, мочевая кислота – 160 мкмоль/л, сахар крови – 4,7 ммоль/л. Характерно также повышение концентрации ферритина сыворотки (1355,4). В общем анализе мочи изменения не значительны.
По ЭКГ от 04.04.16 г.: имелась отрицательная динамика: синусовая тахикардия, ЧСС 120 в минуту. Неполная блокада правой ножки пучка Гиса. Нарушение процессов реполяризации в верхушке и боковой стенки левого желудочка.
По данным рентгенографии органов грудной клетки от 04.04.16 г.: была выявлена двухсторонняя полисегментарная пневмония, двухсторонний гидроторакс. По МСКТ органов грудной клетки выявлены двухсторонний плеврит, гидроторакс, гидроперикард, фиброзные изменения нижней доли слева. По данным ЭХО-КГ от 07.04.16 г. выявили большое количество жидкости в перикарде. Угроза тампонады. Размеры левых отделов у верхней границы нормы. Уплотнение стенок аорты, створок аортального клапана, митрального клапана. Митральная недостаточность I ст. По данным УЗИ органов брюшной полости от 04.04.16 г. определялась гепатоспленомегалия, диффузные изменения печени, поджелудочной железы, селезенки. Свободная жидкость в брюшной полости во всех отделах. В цитологических анализах выпотной жидкости плевральной полости атипичные клетки не обнаружены, общий анализ выявил наличие белка до 24,3 г/л, эритроциты сплошь, положительную пробу Ривальта. В проведенных бактериологических анализах и КУМ трехкратно крови, плевральной жидкости роста микрофлоры и КУМ не обнаружили. При обследовании на маркеры гепатита, УМСС, онкомаркеры получены отрицательные результаты.
Данные результатов обследований при выписке:
По данным ЭХО-КГ от 12.05.16 г. выявлено незначительное количество жидкости в перикарде. Размеры левых отделов у верхней границы нормы. Уплотнение стенок аорты, створок аортального клапана, митрального клапана. Митральная недостаточность I ст.
По МСКТ органов грудной клетки от 13.05.16 г. выявлены признаки двухстороннего плеврита, фиброзные изменения нижней доли слева, свободная жидкость 100 мл слева, 120 мл справа.
УЗИ органов брюшной полости от 13.05.16 г. определялась гепатоспленомегалия, диффузные изменения печени, поджелудочной железы, селезенки. Свободная жидкость в брюшной полости около 150 мл (в сравнении с предыдущими обследованиями отмечается значительное уменьшение).
В клиническом анализе крови отмечается повышение уровня гемоглобина до 111 г/л, тромбоцитопения 64 х10 9 , без геморрагического синдрома, эскалация лейкоцитов достигла 1,3 х10 9 , повышение уровня нейтрофилов, СОЭ – 25мм/ч. В биохимическом анализе отмечается увеличение общего белка до 60 г/л.
Пациент соответствует критериям для постановки данного диагноза: анамнеза, объективных и дополнительных лабораторных и инструментальных методов исследования. С марта 2016 года у пациента отмечается резкое ухудшение состояния, вероятно, спровоцированное перерывом в приеме колониестимулирующего препарата, на фоне чего отмечается ухудшение состояния больного и развитие гемофагоцитарного лимфогистиоцитоза, что подтверждается клиническими и лабораторными данными: длительной лихорадкой, рефрактерной к антимикробной терапии, спленомегалией, отечным синдромом, геморрагическим синдромом, гепатоспленомегалией. [4]. По литературным данным заболевание начинается остро. Триггером клинической манифестации заболевания являются банальные инфекции (особенно герпес, вирусные). Физическое и психомоторное развитие пациента до момента развития гемофагоцитарного синдрома обычно не страдает, как и в случае с данным пациентом. Сохраняется лихорадка, прогрессирует гепатоспленомегалия, появляется и нарастает неврологическая симптоматика, включающая раздражительность, судороги, менингеальные знаки. Лабораторные проявления включают цитопению периферической крови – анемию, тромбоцитопению, лейкопению и нейтропению. Характерно развитие коагулопатии, реже – тотальной гипокоагуляцией, обусловленной печеночно-клеточной недостаточностью. Среди показателей биохимического анализа крови наиболее специфичным является гипертриглицеридемия, развитие которой обусловлено ингибированием липопротеинлипазы. Метаболические аномалии часто включают гипоальбуминемию, гипонатриемию, повышение уровня маркеров гепатоцеллюлярного повреждения: аланин и аспартатаминотрансферазы (соответственно, АЛТ и АСТ) билирубина, лактатдегидрогеназы. Характерно повышение содержания ферритина сыворотки. Все вышеперечисленные данные подтверждаются у наблюдаемого пациента [5].
Заключение. Данный клинический случай интересен в плане наблюдения за пациентом с редким наследственным заболеванием – синдромом Чедиака – Хигаси и достижением пациентом 19-летнего возраста. По-видимому, адекватная и своевременная терапия, наблюдение за пациентом гематологами позволило достичь указанного возраста.
Гемофагоцитарный синдром, развившийся у больного, серьезно осложнил заболевание с неблагоприятным прогнозом. Иммуносупрессивная и сопроводительная терапия не была показана и рекомендована данному пациенту. Существует единственный радикальный метод лечения – аллогенная трансплантация костного мозга от HLA-идентичных родственников или доноров, совместимых по локусу D. В данном случае нашему пациенту было отказано в трансплантации костного мозга в связи с отсутствием доноров-сиблингов и была рекомендована симптоматическая терапия.
Синдром Чедиака-Хигаси

Синдром Чедиака-Хигаси — редкое аутосомно-рецессивное заболевание, которое характеризуется тяжелым врожденным иммунодефицитом по причине дефекта гена CHS1/LYST. Проявляется болезнь частыми бактериальными инфекциями, коагулопатией, нарушениями пигментации кожи, а также прогрессирующими неврологическими расстройствами [1].
Иммунологические нарушения выражаются в нейтропении и нехватке естественных киллеров (NK-клеток). Зачастую заболевание оказывается летальным — большая часть пациентов умирает в детстве. Заподозрить синдром Чедиака-Хигаси можно по тем же признакам, что и любой врожденный иммунодефицит, однако имеется и довольно специфичный критерий — гигантские внутриклеточные включения (или органеллы) в популяциях разных клеток и определенные дефекты в них [2].
Чтобы более наглядно описать происходящие процессы при данном заболевании, еще во второй половине прошлого столетия была предложена оригинальная модель «бежевой мыши». Вследствие мутации в определенном гене (в то время конкретная точка генома, разумеется, не была известна) пигментация экспериментальной мыши изменялась, и исследователи определили ее цвет как «бежевый» («beige»). Помимо цвета у таких животных имелся еще ряд аномалий в виде огромных гранул меланина в лимфоцитах, нейтрофилах и эозинофилах (рис. 1), также подобные включения наблюдались в ряде других клеток [3].
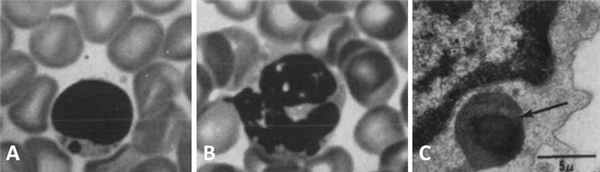
Рисунок 1 | Периферическая кровь бежевой мыши. Световая микроскопия: определяются гранулы в цитоплазме лимфоцита (А) и нейтрофила (В). Электронная микроскопия: объемная гранула с темным содержимым в цитоплазме лимфоцита. Относится к непостоянным аномальным включениям [3].
Позднее данный ген у мыши был идентифицирован и назван Lyst (кодирует регулятор лизосомального транспорта — lysosomal trafficking regulator), человеческий ген обозначают как LYST. Фермент, за экспрессию которого отвечает LYST, обеспечивает нормальный экзо- и эндоцитоз лизосомы; в случае мутации лизосома не может выполнять свои функции [4].
Сегодня этот ген настолько прочно ассоциируется с синдромом Чедиака-Хигаси, что в современной литературе его обозначают как CHS1/LYST(человеческий ген Chediak-Higashi syndrome-1 или LYST — оба варианта равнозначны) [1].
Роль регулятора лизосомального транспорта (LYST)
Данный белок кодируется достаточно объемным геном LYST, который содержит 55 экзонов и расположен в локусе 1q43. Большая длина нуклеотидной последовательности этого гена — фактор неоднозначный: с одной стороны, это существенно затрудняет диагностику, с другой — большое количество мутаций вообще никак не влияет на жизнедеятельность человека и не вызывает синдром Чедиака-Хигаси [5].
Ген кодирует одноименный большой каталитический белок — регулятор лизосомального транспорта (425 kDa, 3801 аминокислотное основание). Протеин относится к семейству BEACH — (Beige and Chediak-Higashi) — и, по-видимому, стал первым его представителем [4]. Белки данного семейства имеют три общих С-концевых домена: PH (Pleckstrin-homology домен), BEACH (Beige and Chediak-Higashi домен) и WD40-повторения (рис. 2) [5].
Рисунок 2 | Строение шести родственных белков семейства BEACH слизевика рода Dictyostelium [6].
На рис. 2 изображена диаграмма, наглядно показывающая общность строения шести белков семейства BEACH (в данном случае — шесть белков слизевика рода Dictyostelium). Зеленым цветом обозначен собственно BEACH-домен, наиболее консервативный во всех белках. «Овалами» у С-конца обозначены WD40-повторения — участки, предположительно вовлеченные в белок-белковые взаимодействия. Другими цветами показаны иные идентичные аминокислотные последовательности [6].
Сегодня известно, что белки данного семейства есть у разных представителей животного мира. Имея примерно одинаковое строение, они выполняют многочисленные и крайне сложные функции, обсуждение которых выходит за рамки данной темы.
Собственно LYST обеспечивает секрецию лизосомальных ферментов. Без белка-регулятора транспорт протеолитических ферментов невозможен, вследствие этого цитотоксическая активность фагоцитарных клеток нарушается. Этим также объясняется нарушение транспорта различных метаболитов (накопление в клетках гранул с меланином, что обеспечивает яркий клинический признак синдрома Чедиака-Хигаси), а также нарушения метаболизма некоторых других белков, что обусловливает клиническую картину заболевания и сопутствующие патологии, точный механизм развития которых все еще неизвестен [1,3].
Клиническая картина
Существует два основных клинических проявления синдрома Чедиака-Хигаси: «парциальный альбинизм» и рецидивирующие пиогенные инфекции. Оба, на первый взгляд, не связанных между собой критерия имеют единую причину — дефект LYST, вследствие чего нарушается транспорт меланина и лизосомальных ферментов.
Цвет волос пациента может быть от сероватого до буквально седого — зависит от этнической принадлежности пациента. Также нарушается пигментация радужной оболочки, следствием чего становится фоточувствительность и сниженная функция зрительного аппарата.
Рецидивирующие бактериальные (в том числе оппортунистические) инфекции часто поражают дыхательные пути, кожу. Возможны дефекты свертывающей системы из-за нарушенного синтеза тромбоцитов, проявляется это небольшими кровоподтеками (зачастую — на слизистых), однако иногда развивается полноценный геморрагический синдром.
Помимо этого возможны неврологические нарушения: атаксия, сенсорные расстройства, прогрессирующая нейродегенерация [1].
Первые проявления заболевания начинаются с раннего детства. Наиболее опасными осложнениями считаются инфекции, для предупреждения которых допустима профилактическая антибиотикотерапия. И все же большинство пациентов умирает в первые десять лет жизни. Наиболее распространенными причинами смерти являются кровотечения, инфекции или гемофагоцитарный лимфогистиоцитоз [7].
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз (ГФЛГ) — патологическое состояние, которое может быть вызвано разнообразными причинами, часто — первичными иммунодефицитами (к числу которых относится и синдром Чедиака-Хигаси). Суть данного состояния — гиперпродукция гистиоцитов и иммунокомпетентных клеток [5].
Как уже было сказано выше, мутация LYST нарушает цитотоксическую функцию клеток, но не метаболизм регуляторных факторов. В ответ на антиген развивается обыкновенная воспалительная реакция — однако элиминация чужеродного агента невозможна. В связи с этим продолжается продукция провоспалительных факторов (ИФН-γ, ФНО, интерлейкины), что увеличивает количество лимфоцитов, нейтрофилов, повышает активность макрофагов. Гиперпродукция цитокинов, не ингибированная по механизму обратной связи, иногда называется «цитокиновым штормом» [5].
Следствием этого является лимфогистиоцитарная инфильтрация различных тканей с развитием в них разнообразных повреждений, а макрофаги могут разрушать нормальные функционирующие клетки (в том числе — форменные элементы крови) [5].
Диагностика синдрома Чедиака-Хигаси и сопутствующих заболеваний
В диагностировании синдрома Чедиака-Хигаси особых сложностей не возникает. Как уже было сказано выше, основная задача клинициста — распознать врожденное иммунодефицитное состояние, дальнейшая диагностика основана на данных иммунограммы и генетического анализа. Достаточно специфичным признаком является накопление больших внутриклеточных везикул в различных клетках, в том числе — лейкоцитах [2].
Одним из наиболее серьезных осложнений при данном синдроме является развитие ГФЛГ. Диагностические критерии при этом патологическом состоянии можно представить следующим образом [5]:
- Наличие генетического дефекта, связанного с ГФЛГ (зачастую это первичные иммунодефициты).
- Наличие как минимум пяти из нижеперечисленных критериев:
- лихорадка;
- спленомегалия;
- цитопения хотя бы в двух клеточных популяциях:
- гипертриглицеридемия (> 3 ммоль/л) или гипофибриногенемия ( < 1,5 г/л);
- гиперферритинемия > 500 мкг/л;
- растворимые молекулы CD25 > 2400 Ед/мл;
- гемофагоцитоз в костном мозге, селезенке, лимфоузлах
- низкая (вплоть до полного отсутствия) цитотоксическая активность NK-клеток.
Важно отметить: ГФЛГ является настолько частым серьезным осложнением, что в современной литературе принято разделять синдром Чедиака-Хигаси на две формы: «классическую» (с развитием ГФЛГ) и «атипичную» (без такового) [5].
Лечение синдрома Чедиака-Хигаси и сопутствующих патологий
Как и в большинстве наследственных иммунодефицитов, вариантов терапии немного. Наиболее распространенным методом лечения врожденных иммунологических нарушений является пересадка гемопоэтических клеток; данный синдром не является исключением.
В 2007 году в «Bone Marrow Transplantation» появилась публикация, авторы которой сообщали о 35 случаях проведения пересадки гемопоэтических клеток пациентам с синдромом Чедиака-Хигаси [8]. Перед проведением операции лечение осуществлялось в лучшем случае патогенетическое, призванное замедлить прогрессирование заболевания, избежать осложнений и подготовить пациента к пересадке гемопоэтических клеток.
Всего 13 пациентов получили материал от HLA-идентичного донора (родного брата или сестры), 10 — от родственника, еще 12 — от несвязанного донора. По результатам проведенной терапии, 27 (77 %) из 35 пациентов достигли ремиссии, однако 5-летняя выживаемость составила 62 % (22 пациента).
Наиболее распространенными причинами смерти были посттрансплантационные осложнения и хронические заболевания. Среди умерших — большинство получило аллотрансплантат от не полностью подходящего донора.
Таким образом, исследователи пришли к выводу, что подобная методика может быть достаточно эффективной, если донор и реципиент являются родственниками. Также следует отметить, что шесть пациентов на момент проведения операции были старше девяти лет, что свидетельствует о существенных успехах в сдерживании и предотвращении развития осложнений у пациентов (одному из реципиентов на момент трансплантации было 19 лет).
На современном этапе пересадка гемопоэтических клеток является наиболее эффективным способом лечения синдрома Чедиака-Хигаси [9]. Вероятно, в будущем станет возможна коррекция непосредственно LYST с помощью генно-инженерных методик (например, подобная терапия уже существует для хронической гранулематозной болезни — другого наследственного иммунодефицитного состояния [10]), однако пока что такой альтернативы для синдрома Чедиака-Хигаси нет.
Часто синдром Чедиака-Хигаси осложняется ГФЛГ — тяжелым состоянием, которое требует немедленного лечения. Среди наиболее эффективных методов терапии в гайдлайне 2004 года описаны химиотерапия и пересадка гемопоэтических клеток [11].
Также на протяжении лечения, до и после него в профилактических целях используется антибиотикотерапия [1,11].
Синдром Чедиака-Хигаси

Медицинский эксперт по заболеванию
Проф. Полина СтепенскиРуководитель отделения ТКМ и иммунотерапии у детей и взрослых клиники «Хадасса»
Стаж: более 24 лет
Запись на прием
Трансплантация костного мозга, выполненная ребенку, больному синдромом Чедиака-Хигаси, в раннем возрасте, позволяет улучшить его состояние и предотвратить возникновение инфекций.
В Израиле данную процедуру блестяще проводит профессор Полина Степенски, известный специалист в области ТКМ при лечении орфанных заболеваний у детей.
Что такое синдром Чедиака-Хигаси
К редким иммунодефицитным заболеваниям, относят синдром Чедиака-Хигаси с генерализованной клеточной дисфункцией. Частота встречаемости болезни колеблется от 1 до 2 случаев на 1 млн. новорожденных детей независимо от пола. Синдром Чедиака-Хигаси вызван генной мутацией, на фоне которой в нейтрофилах накапливаются аномальные цитоплазматические гранулы.
Данный синдром был описан кубинским врачом и серологистом А.М. Чедиаком, а также японским педиатром О. Хигаси. В честь них заболевание и было названо синдромом Чедиака-Хигаси. Генетическая аномалия, унаследованная по аутосомно-рецессивному типу, приводит к уменьшению количества и распаду нейтрофильных лейкоцитов, ответственных за уничтожение вирусных и бактериальных агентов. Во всех клетках костного мозга и периферической крови при синдроме Чедиака-Хигаси обнаруживаются крупные цитоплазматические гранулы. Вследствие этого снижается фагоцитоз, у детей возникают тяжелые рецидивирующие инфекции, развивается периферическая невропатия, а также депигментация глаз, кожи и волос.
Синдром Чедиака-Хигаси у грудных детей протекает с инфекционными рецидивами средней тяжести. В ускоренную фазу течения болезнь переходит у подростков старшего возраста.
Код МКБ 10 — пор международной классификации болезней синдрому Чедиака-Хигаси присвоен код Е70.3
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Причины заболевания
Синдром Чедиака-Хигаси вызван мутацией гена CHS1 (Lyst-протеина), которая приводит к аномалии структур клеточных мембран и образованию гигантских патологических гранул в клетках соединительной ткани периневральной области, циркулирующих лимфоцитах и цитоплазме нейронов. Большинство клинических проявлений синдрома Чедиака-Хигаси обусловлены аномальным распределением лизосомальных ферментов. Сниженная активность внутриклеточного переваривания микробов в фагоцитах и кислородного метаболизма на фоне непостоянного высвобождения лизосомальных ферментов из гигантских гранул способствует частым воспалительным процессам и пиогенным инфекциям.
Симптомы
При синдроме Чедиака-Хигаси наблюдаются такие симптомы и признаки:
- Кожно-глазной альбинизм, при котором нарушается процесс пигментации. Светлая кожа в силу повышенной чувствительности склонна к солнечным ожогам. Также у детей с синдромом Чедиака-Хигаси отмечается фотофобия и слезоточивость.
- Офтальмологические расстройства: нистагм (непроизвольные движения глазных яблок), нервный тик, косоглазие, усиленный мигательный рефлекс.
- Инфекции мочевыводящих путей с появлением болезненных, частых и/или затрудненных мочеиспусканий. При этом в моче может быть примесь крови, гноя.
- Расстройства ЖКТ, при которых возникает лихорадка, рвота, тошнота, диарея и запоры.
- Сенсорная и моторная невропатия (мышечная слабость, судороги, парезы, атаксия, нарушение чувствительности).
- Аллергические реакции, в том числе гайморит, синусит, ринит, приступообразный кашель.
- Прогрессирование анемии при синдроме Чедиака-Хигаси вызывает у детей бледность кожи и слизистых оболочек, а также повышенную утомляемость, сонливость и раздражительность.
- Поражения кожи: отечности, шелушения, зуд, высыпания, формирования пустул, язв.
- Задержка умственного развития.
Кроме того, у детей с синдромом Чедиака-Хигаси отмечается повышенная восприимчивость к различным бактериям и грибам. Инфекционные заболевания вызывают желтуху, судороги, отеки, увеличение лимфоузлов, сопровождаются гипертермией, гепатоспленомегалией, коагулопатией. Учащенные вирусные и бактериальные инфекции, а также осложнения пораженных органов и развитие злокачественных очагов приводит к гибели ребенка в течение 1,5-3 лет.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Диагностика
При подозрениях на синдром Чедиака-Хигаси в первую очередь устанавливается наличие подобных генетических болезней у родителей и родственников, изучается анамнез. Затем проводится физикальный осмотр, позволяющий выявить увеличение печени и лимфоузлов, специфические изменения на фоне бактериальных осложнений, оценить физическое развитие. Второй этап комплексного обследования включает лабораторные и инструментальные методы:
- Генетические тесты на наличие мутаций гена LYST (ДНК-диагностика).
- Общий и биохимический анализ крови на почечные ферменты и гигантские гранулы, количество лейкоцитов, тромбоцитов, эритроцитов.
- Иммунологические исследования.
- Рентгенография грудной клетки.
По показаниям врачи могут назначить УЗИ и МРТ печени, почек и других органов. Пациентам с синдромом Чедиака-Хигаси требуется осмотр и консультация врачей разных меднаправлений, в том числе офтальмолога, гематолога, дерматолога и др.
Лечение
Тактика лечения разрабатывается в каждом случае индивидуально, в зависимости от клинической картины, возраста ребенка. При синдроме Чедиака-Хигаси специалисты применяют методы симптоматической терапии, в частности назначают антибиотики различных групп, глюкокортикостероиды, высокие дозы аскорбиновой кислоты и другие препараты для устранения симптоматики патологических процессов, вызванных синдромом Чедиака-Хигаси.
По мере прогрессирования анемии при синдроме Чедиака-Хигаси выполняют процедуру вливания эритроцитарной массы или плазмы. Для кратковременной ремиссии в некоторых случаях необходима спленэктомия (удаление селезенки). С целью нормализации иммунологических и кроветворных функций, а также увеличения количества лимфоцитов, проводят аллогенную трансплантацию костного мозга (ТКМ).
Меры профилактики
Профилактически мер для предотвращения синдрома Чедиака-Хигаси не существует, поскольку это наследственная патология. Семейным парам, которые планируют деторождение и относятся к группе риска, следует пройти генетические исследования.
Возможные осложнения и прогноз
При синдроме Чедиака-Хигаси в целом прогноз неблагоприятный. В большинстве случаев дети не доживают до подросткового возраста по причине тяжелых осложнений, таких как инфильтрация лимфоузлов, печени и селезенки, быстрая пролиферация лимфоцитов. Значительно ухудшают прогностическую картину развитие злокачественных опухолей. После проведения ТКМ при синдроме Чедиака-Хигаси выживаемость в течение последующего 5-летнего периода составляет 55-60%.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Читайте также: