Синдром FXTAS
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Лабораторное исследование для диагностики синдрома тремора/атаксии, ассоциированных с ломкой Х-хромосомой. Тест позволяет определить увеличенное количество CGG-повторов в гене FMR1, что помогает подтвердить предварительно поставленный диагноз.
Приём и исследование биоматериала
Когда нужно сдавать анализ Генодиагностика синдрома тремора/атаксии (FMR1)?
- Подозрение на синдром тремора/атаксии, связанных с ломкой Х-хромосомой;
- Диагностированная патология у кровных родственников;
- Установление возможной причины тремора, атаксии и деменции.
Подробное описание исследования
Синдром FXTAS (тремор/атаксия, связанные с ломкой Х-хромосомой) — наследственное заболевание, которое характеризуется наличием тремора — т.н. «дрожания» некоторых частей тела, — атаксии — расстройства координации движений — и деменции — стойкого снижения познавательных функций. Эти нарушения связаны с мутацией гена FMR1 в структуре Х-хромосомы. Синдром ломкой Х-хромосомы наследуется по материнской линии. Заболеванию в основном подвержены мужчины, клинические симптомы активнее всего проявляются после пятидесяти лет.
Больные с синдромом тремора/атаксии, ассоциированных с ломкой Х-хромосомой, могут иметь следующие характерные клинические симптомы:
- Дрожание конечностей;
- Замедленная речь и движения;
- Чувство онемения и слабость в ногах;
- Недержание мочи;
- Кратковременная потеря памяти;
- Повышенная тревожность.
- Симптомы становятся более заметными в пожилом возрасте.
Хромосомы — специальные содержащиеся в ядре клеток структуры, которые предназначены для хранения, реализации и передачи наследственной информации. Обычно человек наследует от родителей 23 пары хромосом (всего 46), один набор от матери и один от отца. 22 пары хромосом из набора носят название аутосом, а 2 оставшиеся являются половыми хромосомами (Х и Y, которые определяют пол ребенка). У мужчин в наборе одна Х-хромосома и одна Y. У женщин — две Х-хромосомы. Каждая хромосома состоит из плотно упакованных молекул дезоксирибонуклеиновой кислоты (ДНК).
В составе ДНК выделяют участки (сегменты), которые называют генами. Гены состоят из соединенных в определенной последовательности нуклеотидных оснований (аденин — А, цитозин — С, гуанин — G, тимин — Т). Гены являются матрицей для создания (транскрипции) рибонуклеиновой кислоты (РНК). РНК несет в себе информацию для создания белков, регулирующих работу всего организма.
Ген FMR1 кодирует белок FMRP (Fragile X Mental Retardation Protein), необходимый для нормального развития и работы мозга. Он участвует в формировании специальных связей (синапсов) между нервными клетками. Когда возникает мутация гена FMR1, нарушается образование белка FMRP, что приводит к его дефициту или его полному отсутствию.
В гене FMR1 есть участок, который имеет несколько повторов цитозин-гуанин-гуанин (CGG). Этот участок носит название триплет CGG. В норме человек имеет от пяти до сорока четырех таких «повторов». Если «повторов» больше, чем сорок четыре (от 44 до 200), то с возрастом проявляются симптомы заболевания, а женщины имеют очень высокий риск передать потомству данную генетическую мутацию.
Выраженность клинических симптомов связана с количеством CGG-копий — чем их больше, тем выраженнее симптомы. При количестве CGG-повторов более 200 развивается синдром Мартина-Белл, при количестве повторов 55-199 (премутация) развиваются промежуточные формы: синдром тремора/атаксии у мужчин и синдром первичной яичниковой недостаточности у женщин.
Исследование позволяет провести генодиагностику синдрома тремора/атаксии, ассоциированных с ломкой Х-хромосомой, чтобы обнаружить увеличенное количество повторов CGG в гене FMR1.
Также стандартная диагностика основывается на детальном клиническом исследовании больного, анализе минимум трех поколений родословной, в которой особое внимание уделяется заболеваниям среди родственников мужского пола по материнской линии. При этом учитывается то, что мужчины передают мутацию Х-сцепленного гена всем своим дочерям, а женщины – дочерям и сыновьям с вероятностью 50%. Исключающим критерием для Х-сцепленного наследования является факт передачи заболевания от отца к сыну.
Все матери больных тщательно обследуются на наличие клинических признаков патологии, поскольку они часто являются носительницами соответствующих Х-сцепленных мутаций.
Синдром Тремора и Атаксии
Состояние премутации, в отличие от полной мутации, связано с возникновением сверхпродукции матричной РНК, которая, впоследствии, будет иметь негативный эффект на нервную систему. В результате такого высокого уровня мРНК у носителей премутации с возрастом может возникнуть Синдром Тремора и Атаксии (впервые был описан в 2001 году).
Не у всех носителей премутации развивается синдром тремора и атаксии (приблизительно в 20-40%).
FXTAS – это возрастное поздно начавшееся неврологическое заболевание с развитием прогрессирующего тремора при произвольных движениях, нарушением походки и когнитивным снижением.
Заболеванию подвержены, главным образом, пожилые мужчины-носители премутации (старше 50 лет), женщины страдают данным заболеванием реже и симптомы заболевания протекают легче.
Первые признаки заболевания могут проявиться в затруднениях в повседневной жизни, например, возникают трудности с письмом или при использовании столовых принадлежностей, или появляются трудности в удержании равновесия при ходьбе. Так же могут возникать проблемы с кратковременной памятью, наблюдается нарастание общего беспокойства, появление неуместного или импульсивного поведения.
Симптомы FXTAS прогрессируют в течение многих лет или десятилетий, пока выполнение многих ежедневных задач становится не доступно.
Очень часто носителям премутации с FXTAS диагностируют болезнь Паркинсона, старческое слабоумие или болезнь Альцгеймера. Поэтому проблема дифференциальной диагностики FXTAS от схожих форм нейродегенеративных заболеваний на сегодняшний день является актуальной.
Основные симптомы FXTAS
- Интенционный тремор: возникновение дрожания в момент целенаправленного движения руки к объекту, например, при использовании столовых приборов или письменных принадлежностей. В состоянии покоя тремор не столь очевиден.
- Атаксическая походка: походка становится неуверенной, с широко расставленными ногами, наблюдаются проблемы в удержании равновесия, которые могут включать в себя эпизоды падение, а также необходимость поддержки при ходьбе по неровной местности или при хождении вверх / вниз по лестнице.
- МРТ–исследование при FXTAS. Данные МРТ включают в себя «поражение белого вещества в перивентрикулярной области, подкорковых областях и в районе средних ножек мозжечка билатерально».
- Выводы невропатологов о так называемых «FXTAS включения» в клетках головного мозга.
Вторичные симптомы FXTAS
- Паркинсонизм (тремор покоя).
- Проблемы с кратковременной памятью. Это бывает трудно определить, так как с возрастом происходит естественное ухудшение кратковременной памяти. Однако, при FXTAS это может происходить быстрее, чем обычно, или может быть более выраженным, например, человек забывает, что только что съел, сказал или сделал.
- Проблемы с «исполнительными функциями» и принятием решений. Исполнительные функции включают в себя возможность инициировать и завершить деятельность, адаптировать и изменять поведение по мере необходимости и в соответствии с ситуацией, прогнозировать и планировать новые задачи и ситуации. Исполнительные функции позволяют предвидеть результаты, решать проблемы, обобщать и переносить опыт из одной ситуации в другую.
- МРТ, которые являются более общими, чем те, которые перечислены выше, упоминается как «поражение белого вещества головного мозга»
Другие симптомы FXTAS (не считаются официальными диагностическими критериями, но часто встречаются у людей с FXTAS)
- Невропатия или онемение / покалывание в конечностях.
- Нестабильность в настроении, раздражительность, вспышки гнева, личностные изменения.
- Когнитивное снижение: потеря раннее приобретенных навыков, включая математику, чтение и т.д.
- Импотенция, потеря контроля над рефлексами мочевого пузыря или функций кишечник (так называемые проблемы «вегетативной дисфункции»).
- Гипертония, заболевания щитовидной железы, фибромиалгии (чаще встречается у женщин).
Рекомендация для дальнейших диагностических мероприятий с целью подтверждения синдрома FXTAS необходима для:
- Лиц с проявлениями «основных» клинических симптомов (№ 1 или № 2 из группы «основные симптомы FXTAS») и один «основной» симптом по МРТ-данным (№3 или № 4 из группы «основные симптомы FXTAS»)
- Любого человека с наличием «FXTAS включений» на основе неврологических данных.
Часто встречаемые симптомы у женщин с FXTAS.
Женщины также могут страдать в той или иной степени неврологическими симптомами, что и мужчины, но почти всегда с меньшей тяжестью, благодаря присутствию второй Х хромосомы. В целом, для женщин характерна склонность к выраженной тревоге и депрессиям.
Дополнительные симптомы, которые могут наблюдать у некоторых женщин с FXTAS, включают в себя:
Синдром FXTAS (тремор/атаксия, ассоциированные с ломкой Х-хромосомой) (FXTAS)
Синдром хрупкой Х-хромосомы тремор/атаксия - это генетическое заболевание, поражающее преимущественно мужчин, которое вызывает тремор, атаксию и деменцию. Тремор является распространенным ранним симптомом, который сопровождается атаксией, паркинсонизмом, и в конечном счете деменцией. Диагноз подтверждают генетическим тестированием. Тремор часто удается купировать примидоном, пропранололом и/или противопаркинсоническими препаратами.
Синдромом тремора/атаксии, связанным с ломкой Х-хромосомой (FXTAS), страдает примерно 1 мужчина из 3 000 старше 50 лет. Это является результатом премутации (от 50 до 200 повторов аминокислотной последовательности CGG) в Х-хромосоме, в так называемом гене умственной отсталости ломкой Х-хромосомы (FMR1). Синдром хрупкой Х-хромосомы Синдром ломкой Х-хромосомы Синдром ломкой X-хромосомы является генетическим нарушением в X-хромосоме, ведущим к умственной отсталости и нарушениям поведения. Диагностика проводится с помощью молекулярного анализа ДНК. Прочитайте дополнительные сведения , наиболее распространенный вариант умственной отсталости у мужчин, развивается при полной мутации ( > 200 повторов).
Люди с премутацией считаются носителями. Дочери (но не сыновья) мужчин с премутацией наследуют ее. Дети дочерей (внуки/внучки мужчин – носителей премутации FXTAS) с вероятностью 50% наследуют премутацию, которая может трансформироваться в полноценную мутацию при передаче от матери к ребенку (приводя, таким образом, к развитию синдрома ломкой Х-хромосомы).
Синдром FXTAS развивается примерно у 30% мужчин с премутацией и менее чем у 5% женщин с премутацией.
С возрастом риск развития синдрома FXTAS увеличивается.
Симптомы и признаки синдрома FXTAS
Симптомы FXTAS становятся заметными в пожилом возрасте. Чем больше число CGG-копий, тем тяжелее протекает заболевание и раньше появляются его симптомы.
У пациентов с другими нарушениями, ассоциированными с Х-хромосомой (например, синдром Клайнфельтера Синдром Клайнфельтера (47, XXY) Синдром Кляйнфельтера подразумевает наличие двух или более Х-хромосом плюс одну Y-хромосому, что приводит к формированию мужского фенотипа. Диагноз ставят на основании клинических данных и цитогенетических. Прочитайте дополнительные сведенияЗдравый смысл и предостережения
Диагноз FXTAS обсуждают у пациентов с тремором, если на его фоне развиваются признаки атаксии или паркинсонизма.
Деменция начинается с потерей краткосрочной памяти, замедления темпа мышления и трудностей при решении задач. Не исключается также развитие депрессии, тревожности, раздражительности, агрессивности и эмоциональной лабильности.
Ожидаемая продолжительность жизни после появления двигательных нарушений составляет от 5 до 25 лет.
У женщин с премутацией симптомы заболеваний, как правило, менее выражены, что, возможно, связано с компенсаторным действием другой Х-хромосомы. У таких женщин имеется повышенный риск развития бесплодия, нарушения функции яичников, а также раннего развития менопаузы.
Диагностика синдрома FXTAS
Заподозрив FXTAS, нужно спросить пациента, страдает ли кто-либо из его внуков интеллектуальными нарушениями, и отмечались ли у их дочерей ранняя менопауза или бесплодие.
Кроме того, если у пациента имеется синдром ломкой Х-хромосомы, врачи должны определить, есть ли у его бабушки и дедушки симптомы, указывающие на этот синдром; если это так, для детей и внуков таких бабушки и дедушки рекомендуется генетическое консультирование.
Проводят МРТ; этот метод помогает выявить типичное повышение интенсивности сигнала от средних ножек мозжечка.
Диагноз FXTAS подтверждают генетическим тестированием.
Лечение синдрома FXTAS
Примидон, пропранолол, топирамат, габапентин, прегабалин, бензодиазепины и/или противопаркинсонические препараты
Справочные материалы по лечению
1. Hall DA, Berry-Kravis E, Hagerman RJ, et al: Symptomatic treatment in the fragile X-associated tremor/ataxia syndrome. Mov Disord 21 (10):1741–1744, 2006. doi: 10.1002/mds.21001
2. Cabal-Herrera AM, Tassanakijpanich N, Salcedo-Arellano MJ, Hagerman RJ: Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathophysiology and clinical implications. Int J Mol Sci 21 (12): 4391, 2020. Published online 2020 Jun 20. doi: 10.3390/ijms21124391
Ключевые моменты
Синдром FXTAS тремор/атаксия, ассоциированные с ломкой Х-хромосомой поражает примерно 1/3000 мужчин > 50 лет; синдром ломкой X-хромосомы является наиболее распространенной причиной умственной отсталости у мужчин, которая развивается из-за мутации соответствующего гена.
Нужно спрашивать пациентов о том, страдает ли кто-либо из его внуков интеллектуальными нарушениями, и отмечались ли у их дочерей ранняя менопауза или бесплодие, а также спрашивать прародителей пациентов с синдромом ломкой Х хромосомы, были ли у них симптомы, предполагающие FXTAS.
Для подтверждения диагноза необходимо сделать генетическое исследование.
Тремор лечат при помощи примидона, пропранолола, топирамата, габапентина, прегабалина, бензодиазепинов и/или противопаркинсонических препаратов.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Синдром ломкой Х-хромосомы с тремором/атаксией, FMR1, ч. м. (Fragile X-associated tremor/ataxia syndrome (FXTAS), FMR1 gene)
Синдром тремора/атаксии (СТА), ассоциированный с синдромом ломкой Х-хромосомы – это нейродегенеративное заболевание, вызываемое предэкспансией CGG-тринуклеотидных повторов в 5-нетранслируемой области гена FMR1. Заболевание чаще встречается у мужчин и клинически проявляется примерно на седьмом десятилетии жизни.
В норме количество повторов CGG составляет менее 54. При экспансии CGG-тринуклеотидных повторов более 200 наблюдается уменьшение количества или полное отсутствие FMR протеина, что приводит к развитию синдрома ломкой X-хромосомы (синдром Мартина-Белл), который является одной из самых распространенных наследственных причин умственной отсталости. При синдроме тремора/атаксии число тринуклеотидных повторов находится в пределах 55-199 и синтезируется нормальное количество белка FMR, однако формирующаяся мРНК избыточно накапливается в лейкоцитах и головном мозге. Высокий уровень мРНК оказывает токсическое действие и связан с развитием клинической картины.
К клиническим проявлениям относятся мозжечковая атаксия, акционный тремор, паркинсонизм, когнитивные нарушения. Также отмечаются варьирующие в степени выраженности психиатрические нарушения, периферическая нейропатия, вегетативная дисфункция. Характерная МРТ-картина характеризуется наличием очагов в средней ножке мозжечка или ствола мозга, в белом веществе, а также генерализованной атрофии головного мозга. У женщин вследствие наличия второй X-хромосомы данный синдром не развивается, однако известно, что носительницы чаще страдают от депрессии и тревожных расстройств. Также у них отмечается повышенный риск развития первичной яичниковой недостаточности.
Синдром Мартина-Белл является Х-сцепленным и наследуется доминантно, то есть имеется 50% риск передачи заболевания потомству от больной матери. Необходимо отметить, что при синдроме тремора/атаксии клиническая картина коррелирует с размером премутации. Также наблюдается увеличение пенетрантности с увеличением возраста пациента и в зависимости от уровня премутации.
Согласно рекомендациям, определение премутации в гене FRM1 необходимо для подстановки диагноза синдрома тремор/атаксии, ассоциированный с синдромом ломкой Х-хромосомы. Диагностические критерии также включают клинические и радиологические признаки.
Специальной подготовки не требуется. Взятие крови желательно проводить не ранее 4 часов после приема пищи.
- при наличии мозжечковой атаксии, акционного тремора, деменции неизвестного генеза у пациентов старше 50 лет;
- наличие в семье случаев аутизма, задержки развития, когнитивных нарушений неизвестного генеза;
- семейный анамнез синдрома Мартина-Белл или синдрома тремора/атаксии.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
| Количество CGG-повторов | Диагноз и прогноз |
| 5-44 норма | Синдром тремора/атаксии исключен, низкий риск его возникновения у потомков. |
| 45-54 умеренное увеличение | Синдром тремора/атаксии исключен, однако имеется риск развития синдрома у родственников. |
| 55-199 предэкспансия | Повышен риск развития синдрома тремора/атаксии, ассоциированного с синдромом ломкой Х-хромосомы, необходима оценка клинических и радиологических признаков. Также имеется риск развития синдрома Мартина-Белл у потомков. |
| ≥200 выраженная экспансия | Синдром Мартина-Белл подтвержден. Риск передачи от матери потомкам составляет 50%. |
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации. ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
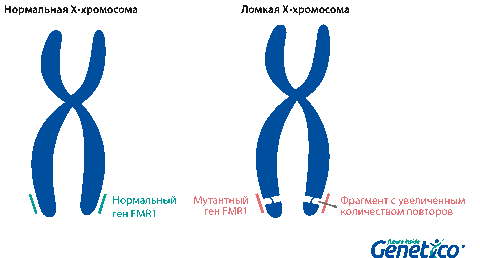
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Медико-генетический центр и лаборатория
Широкий спектр услуг в области медицинской генетики
Читайте также: