Синдромы Дубина-Джонсона, Ротора у новорожденного - клиника, диагностика
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Желтуха у новорожденных при нарушениях обмена веществ
Младенцы с тирозинемией (будет рассмотрена детально далее), галактоземией и фруктозурией (наследственной непереносимостью фруктозы) имеют схожие клинические проявления, особенно в первые дни и недели после рождения. Часто доминируют выраженная желтуха, гепатоспленомегалия, нарушения коагулограммы крови и ухудшение состояния ребенка в динамике.
У детей с галактоземией симптомы заболевания могут быть менее выражены, но по прошествии нескольких месяцев развиваются катаракта, цирроз печени и задержка психомоторного развития. Подобная ситуация может быть и у ребенка с тирозинемией, когда острая фаза заболевания отсутствует и лишь через несколько месяцев диагностируют цирроз, рахитические изменения костей и заболевание почек. Среди детей с этим заболеванием отмечена высокая частота опухолей печени.
Холестаз в сочетании с рвотой, отвращением к сладкой пище и фруктозурией, наиболее вероятно, является следствием непереносимости фруктозы. Каждое из указанных метаболических заболеваний может быть причиной дисфункции почек, которая проявляется аминоацидурией, глюкозурией и фосфатурией (синдром Фанкони). Более точная их диагностика основана на проведении специфических тестов на переносимость отдельных субстанций и определении активности фермента в эритроцитах (для галактоземии), печени или почках (в случае подозрения на непереносимость фруктозы), а также обнаружении сукцинилацетона в моче (при тирозинемии). Для начального скрининга подходит исследование мочи на нередуцирующие сахара, органические кислоты и аминокислоты.
Персистирующая желтуха нетипична для младенцев с муковисцидозом, но она может наблюдаться, если это заболевание сопровождается мекониальным илеусом, гиперчувствительностью к лекарствам или частичной обструкцией общего желчного протока в результате сгущения желчи, а также если ребенок находится на парентеральном питании.

Дефицит а1-AT является наиболее частым нарушением метаболизма при патологии печени у младенцев. Обычно заболевание манифестирует развитием холестаза в сочетании с желтухой и гепатомегалией.
Диагноз подтверждается низким уровнем а1-АТ в сыворотке и определением ингибиторов протеазы (Pi) белка. Пока не существует специфического лечения, но считается перспективным использование генной терапии в будущем. Важно выявлять детей группы риска и проводить соответствующее генетическое консультирование в семьях. Согласно принятой тактике трансплантацию печени проводят детям более старшего возраста в терминальной стадии болезни печени, вызванной дефицитом а1-АТ. Прогноз при этом заболевании очень разнообразен.
Несмотря на наличие умеренно выраженной гепатоцеллюлярной дисфункции, через несколько месяцев после рождения может наступить клиническое улучшение. У некоторых детей в более старшем возрасте исходом заболевания могут стать билиарный цирроз и портальная гипертензия.
Наследственные болезни накопления, такие как болезнь Нимана-Пика и болезнь Гоше, обычно приводят к гепатоспленомегалии у младенцев; холестаз для этих болезней не типичен. Болезни накопления встречаются редко, поэтому не следует проводить их диагностику у детей с холестатической желтухой на начальном этапе диагностического поиска.
Полное парентеральное питание, как правило, приводит к поражению печени у новорожденных. ППП-ассоциированная гепатопатия обычно проявляется в виде холестаза, который проходит после отмены парентерального питания. У некоторых детей процесс восстановления печени может занять несколько месяцев, у других может развиться стойкое нарушение ее функции или печеночная недостаточность, приводящая к необходимости трансплантации печени.
Частыми факторами, влияющими на восстановление функции печени, являются сопутствующие инфекции (например, сепсис), хирургическая патология ЖКТ и недоношенность. Наличие этих факторов обусловливает недостаточную изученность патогенеза гепатопатии, ассоциированной с ППП. Основное лечение этого состояния заключается в отмене парентерального питания и разумном назначении энтерального кормления.
Врожденный стридор
Врожденный стридор – патологическое шумное (свистящее или шипящее) дыхание, обусловленное врожденной аномалией строения гортани или трахеи. Врожденный стридор проявляется громким затрудненным дыханием, которое увеличивается при плаче, кашле. Диагностика врожденного стридора проводится педиатром, отоларингологом и пульмонологом с учетом данных анамнеза, ларингоскопии, трахеобронхоскопии, эзофагоскопии. Лечения врожденного стридора, обусловленного функциональными причинами, не требуется: по мере роста хрящей гортани (к 2-3 годам) стридорозное дыхание исчезает. Если патология связана с органическими причинами, может потребоваться хирургическое вмешательство.
Общие сведения
Врожденный стридор является заболеванием детей раннего возраста, с которым приходится сталкиваться специалистам в области педиатрии, отоларингологии и пульмонологии. Врожденный стридор обычно проявляется с рождения, реже дебютирует на первом месяце жизни ребенка или несколько позже. Характерным признаком стридора служит патологически шумное дыхание, связанное с преодолением сопротивления, которое встречается на пути воздушных масс при прохождении через суженный участок гортани. Врожденный стридор не является самостоятельной нозологической формой, а указывает на дыхательную обструкцию, обусловленную различными патологическими состояниями гортани и трахеи.
Причины врожденного стридора
От 60 до 70% случаев врожденного стридора у детей обусловлены аномалиями развития гортани и верхних дыхательных путей. Чаще всего стридорозное дыхание обусловлено врожденной слабостью наружного кольца гортани (ларингомаляцией). В этом случае во время вдоха надгортанник и черпалонадгортанные складки пролабируют в полость гортани, что сопровождается ее обструкцией и стридорозным звуком на вдохе. Ларингомаляция часто наблюдается у детей, рожденных от преждевременных родов, страдающих гипотрофией, рахитом, спазмофилией. В ряде случаев врожденная слабость гортани может сочетаться с другими пороками дыхательных путей – например, трахеомаляцией и трахеобронхомаляцией.
Данные нарушения связывают с локальной формой мышечной гипотонии, обусловленной задержкой развития нервно-мышечного аппарата. У детей с врожденным стридором нервно-мышечная недостаточность также может проявляться гастроэзофагеальным рефлюксом, ахалазией кардии, апноэ сна, птозом век.
Значительно реже среди причин врожденного стридора выявляются доброкачественные опухоли гортани (гемангиомы, лимфангиомы, папилломатоз гортани), злокачественные новообразования, атрезия хоан, парезы и параличи голосовых складок, рубцовый ларинготрахеальный стеноз, врожденный зоб и др. Если врожденный стридор возникает во время кормления, необходимо думать о пищеводно-трахеальном свище или расщелине гортани.
Врожденный стридор может встречаться как изолированно, так и входить в структуру генетических синдромов (Дауна, Марфана, Пьер–Робена и др.). Нередко врожденному стридору сопутствует другая врожденная патология - гипертензионно-гидроцефальный или судорожный синдром, пороки сердца (открытое овальное окно, дополнительная хорда желудочка), легочная гипертензия и др.
Приобретенный стридор может являться признаком опухолей трахеи и бронхов, обтурации трахеобронхиального дерева инородными телами, бронхиальной астмы, аллергического отека дыхательных путей, инфекций (эпиглоттита, заглоточного абсцесса, крупа), подсвязочного стеноза, обусловленного длительной интубацией, раком гортани или пищевода, тиреоидита.
Классификация врожденного стридора
По степени нарушения дыхания врожденный стридор классифицируют следующим образом:
- I степень (компенсированная) - врожденный стридор не требует лечения;
- II степень (погранично-компенсированная) - врожденный стридор требует динамического наблюдения и, возможно, лечения;
- III степень (декомпенсированная) – врожденный стридор, требующий лечения;
- VI степень – врожденный стридор, несовместимый с жизнью, требующий немедленных реанимационных мероприятий и хирургического лечения.
Различают три типа врожденного стридора: инспираторный, экспираторный и двухфазный. Инспираторный стридор развивается на вдохе; при этом образуется стридорозный шум низкого звучания, обусловленный локализацией поражения выше голосовых складок (в области гортаноглотки или верхнего отдела гортани). При двухфазном стридоре имеет место обструкция дыхательных путей на уровне голосовых складок; при этом возникает шумное дыхание высокого звучания. Врожденный стридор экспираторного типа, развивающийся на выдохе, обусловлен обструкцией ниже голосовых складок и характеризуется средней высотой стридорозного звука.
Симптомы врожденного стридора
Врожденный стридор проявляется уже вскоре после рождения ребенка и усиливается в первые недели жизни. При этом обращает внимание характерное слышимое на расстоянии громкое звучание, возникающее при прорыве воздушной струи через суженную гортань. Шум может быть свистящим, звонким или шипящим, глухим; напоминать воркование голубей, мурлыканье кошки, петушиный крик или кудахтанье курицы. Во время сна, нахождения в теплом помещении, в состоянии покоя интенсивность шума снижается; при сосании, плаче, кашле – напротив, увеличивается.
В большинстве случаев врожденный стридор протекает в легкой, компенсированной степени, позволяя ребенку расти и развиваться нормально. При компенсированном врожденном стридоре физическое состояние ребенка, как правило, не страдает, акт сосания не нарушен, голос сохранен. Однако в некоторых случаях, стридорозное дыхание может сочетаться с явлениями дисфонии и дисфагии.
При возникновении ОРВИ, сопровождающихся катаральными изменениями верхних дыхательных путей, возникает острый приступ стридора. Это проявляется одышкой, ларингоспазмом, цианозом кожных покровов, втяжением межреберных промежутков грудной клетки, яремной ямки и эпигастральной области при вдохе. При тяжелой форме врожденного стридора может развиться асфиксия и острая дыхательная недостаточность.
Наличие у ребенка врожденного стридора может способствовать развитию ларингита, трахеита, бронхита, тяжелой пневмонии, бронхиальной астмы.
Диагностика
Для выяснения причин врожденного стридора необходим осмотр ребенка педиатром, отоларингологом, пульмонологом, неврологом, гастроэнтерологом. При осмотре оценивается общее состояние ребенка, ЧД и ЧСС, окраска кожных покровов, участие вспомогательной мускулатуры в акте дыхания, втяжение уступчивых участков грудной клетки при дыхании и т. д.
- Для визуальной оценки состояния гортани проводится микроларингоскопия.
- При необходимости выполняется рентгенография гортани и мягких тканей шеи, УЗИ гортани, КТ и МРТ, рентгенография грудной клетки, трахеобронхоскопия, бронхография.
- При сочетании симптомов врожденного стридора с дисфагией обследование дополняется рентгеноскопией пищевода, эзофагогастродуоденоскопией, УЗИ брюшной полости.
- При дисфонии и афонии целесообразно проведение нейросонографии, ЭЭГ, КТ головного мозга.
- При подозрении на врожденный зоб необходима консультация эндокринолога с проведением УЗИ щитовидной железы, определением уровня ТТГ, Т3, Т4.
Дифференциальную диагностику врожденного стридора следует проводить с ларингитом, истинным и ложным крупом, заглоточным абсцессом, инородными телами гортани, трахеи и бронхов, бронхоаденитом при туберкулезе, лимфогранулематозом.
Лечение врожденного стридора
В большинстве случаев явления компенсированного и погранично-компенсированного врожденного стридора, уменьшаются к 6 месяца жизни ребенка и исчезают полностью к 2-3 годам. Рекомендуется регулярное наблюдение отоларинголога; специального лечения при этом обычно не проводится.
В ряде случаев при ларингомаляции прибегают к нанесению лазером надрезов на надгортанник, рассечению черпалонадгортанных складок либо удалению части черпаловидных хрящей. При острых приступах врожденного стридора, развившихся на фоне ОРВИ, требуется немедленная госпитализация. В стационаре могут назначаться гормональные препараты, ингаляции, бронхолитики. При развитии критического состояния показана трахеотомия или интубация с ИВЛ.
При врожденном стридоре, обусловленном опухолевыми процессами, необходимо эндоскопическое удаление доброкачественных новообразований гортани. При стридоре, вызванном врожденный гипотиреозом, проводится гормонозаместительной терапия.
Прогноз и профилактика осложнений при врожденном стридоре
По мере роста ребенка, хрящи гортани становятся тверже, просвет гортани - шире, поэтому явления врожденного стридора могут спонтанно регрессировать к 2-3 годам. В этот период необходимо заботиться о профилактике простудных заболеваний, полноценном питании, проведении закаливающих мероприятий, создании благоприятной психологической обстановки. При наличии органических причин врожденного стридора и сопутствующей патологии необходимо их своевременное устранение.
В случае присоединения респираторной инфекции, декомпенсации врожденного стридора и развитии дыхательной недостаточности, прогноз может вызывать опасения.
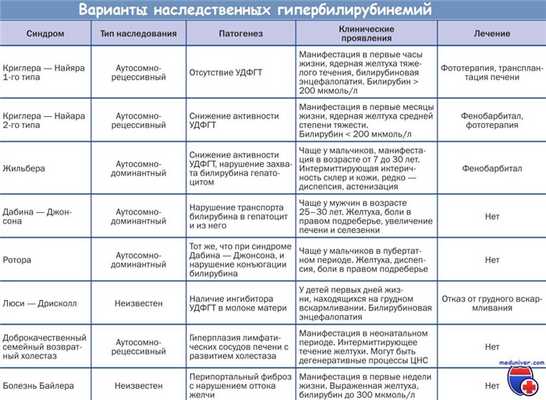
Синдром Дабина-Джонсона ( Генетически обусловленный пигментный гепатоз , Энзимопатическая желтуха )
Синдром Дабина-Джонсона — это хроническое наследственное заболевание, которое характеризуется нарушением выделения билирубина из гепатоцитов в желчь. Основное клиническое проявление — интермиттирующая желтуха. Для болезни также характерны диспепсические расстройства, снижение аппетита и ухудшение общего самочувствия. Диагностика синдрома Дабина-Джонсона включает биохимические анализы крови и мочи, бромсульфалеиновую пробу, инструментальные методики (УЗИ, лапароскопию, биопсию печени). Лечение предполагает коррекцию образа жизни, назначение щадящей диеты. При необходимости используют желчегонные препараты и другие лекарственные средства.
МКБ-10
Синдром имеет несколько синонимов — энзимопатическая желтуха, генетически обусловленный пигментный гепатоз. Нозологическая форма названа в честь двух американских ученых — И.Н. Дабина и Ф.Б. Джонсона, описавших синдром в 1954 году. Это редкое заболевание, в 70% проявляющееся в молодом возрасте. В основном синдром Дабина-Джонсона наблюдается у жителей Среднего Востока. Наибольшая частота встречаемости среди иранских евреев — 1 случай на 1300 населения. У 60% больных патология сопровождается снижением активности факторов коагуляции и кровотечениями. Заболеваемость не зависит от пола.
Причины
Синдром имеет генетическую природу, характеризуется аутосомно-рецессивным путем наследования. Среди родственников болезнь может проявляться как у мужчин, так и у женщин, повторяется через 1-2 поколения. Наследственный дефект представлен мутацией в нуклеотидной последовательности, обеспечивающей кодирование белка MRP2. Этот протеин отвечает за экскрецию (выделение) конъюгированного билирубина и органических анионов в желчные ходы.
Патогенез
Вследствие дисфункции АТФ-зависимой транспортной системы канальцев желчь не проходит в желчные капилляры, из-за чего билирубин накапливается в печеночных тканях. Длительно существующий синдром сопровождается обратными биохимическими реакциями: избыточное количество прямого билирубина освобождается от глюкуроновой кислоты. Полученный непрямой билирубин также проникает в кровоток, обуславливает токсическое действие на нервную систему.
Нарушается выделение активных метаболитов эпинефрина (триптофана, тирозина). Как следствие, в печени накапливаются меланиноподобные пигменты, расположенные в лизосомах гепатоцитов. При макроскопическом исследовании в печеночной паренхиме видны множественные темные пятна — так называемая «шоколадная печень». При исследовании образцов под микроскопом наблюдаются скопления пигментных зерен большей частью в центре долек.
Симптомы
Обычно манифестация синдрома Дабина-Джонсона происходит в 20-30-летнем возрасте, хотя первые характерные признаки появляются уже у подростков. Крайне редко заболевание выявляется у детей. У женщин с бессимптомным течением энзимопатической желтухи клинический дебют болезни могут спровоцировать наступление беременности или прием контрацептивных средств.
Основный симптом заболевания — желтуха, не сопровождаемая кожным зудом. Сначала возникает желтушность склер и слизистых оболочек, затем кожа также приобретает желтушный оттенок. Желтуху могут усиливать изнурительные физнагрузки, стрессовые ситуации, интеркуррентные инфекции. Как правило, желтушные периоды сменяются безжелтушными, хотя у ряда пациентов иктеричность кожи сохраняется постоянно.
Синдром периодически обостряется. Пациент жалуется на сильные боли справа в подреберье, реже — в околопупочной области. Иногда боль отдает в правое плечо или лопатку. Болевой синдром может быть настолько интенсивным, что напоминает печеночную колику. Одновременно с болью беспокоят тошнота, горечь во рту. Изредка бывает рвота, которая не приносит облегчения. Общая интоксикация проявляется повышенной утомляемостью, сонливостью, снижением аппетита.
Осложнения
Синдром Дабина-Джонсона отличается доброкачественным течением и при правильном лечении не вызывает неприятных последствий. Частое осложнение при длительно протекающем заболевании — воспалительные процессы в желчном пузыре и протоках. Воспаление может переходить и на печеночную паренхиму, вызывая холестатический гепатит. При наличии этих патологий желтуха у больных синдромом Дабина-Джонсона становится постоянной.
При присоединении бактериальной инфекции может возникать эмпиема желчного пузыря и гнойный холангит, которые при неблагоприятных условиях трансформируются в ограниченный перитонит. У пожилых людей наследственный пигментный синдром провоцирует появление фиброзных изменений в печеночных дольках. В результате снижается функциональная активность печени. Вследствие нарушения синтеза факторов свертывания крови возрастает риск кровотечений.
При физикальном обследовании пациентов врач-гастроэнтеролог или гепатолог пальпирует выступающий на несколько сантиметров из-под реберной дуги край печени. Синдром дифференцируют с болезнью Ротора, гепатитами, раковыми метастазами в печень. Симптомы поражения желчного пузыря (Мюсси, Ортнера, Кера) отрицательные. Для подтверждения заболевания используют следующие лабораторные и инструментальные исследования:
- Анализы крови. Основным диагностическим критерием синдрома является повышение уровня общего билирубина более 85 мкмоль/л, при этом массовая доля прямого билирубина превышает 15%. При выполнении коагулограммы у 60% пациентов выявляют снижение активности протромбина и уменьшение протромбинового времени.
- Анализ мочи. В моче обнаруживают повышенный уровень билирубина. Для дифференциальной диагностики с близким по клинической симптоматике синдромом Ротора оценивают соотношение копропорфиринов I и III типа в моче. При гепатозе Дабина-Джонсона количество копропорфирина 1 типа составляет 80%, а 3 типа — 20%.
- Бромсульфалеиновая проба. Анализ наиболее чувствителен для оценки печеночных функций. При синдроме Дабина-Джонсона спустя 45 минут концентрация в крови специального красителя, который предварительно ввели внутривенно, составляет более 6%. Такая проба считается положительной и указывает на снижение поглотительно-выделительной функции печени.
- УЗИ органов брюшной полости. При сонографии синдром проявляется увеличением печени на 1-2 см, у пациентов среднего и пожилого возраста нередко обнаруживают очаги фиброза. Размеры и контуры желчного пузыря не изменены, конкременты не визуализируются. Характерно одновременное увеличение селезенки.
- Инвазивные методы. В сомнительных ситуациях показана диагностическая лапароскопия, которая позволяет врачу осмотреть наружную поверхность печени и выявить характерные коричневые пятна. Чтобы подтвердить диагноз, назначается чрескожная биопсия печеночной ткани с последующим осмотром биоптатов под электронным микроскопом.
Лечение синдрома Дабина-Джонсона
Этиопатогенетическое лечение заболевания не разработано. Главная роль в предупреждении ухудшений состояния больных принадлежит немедикаментозным мероприятиям. Чтобы предотвратить обострения синдрома Дабина-Джонсона, пациентам рекомендуют по возможности избегать физического переутомления и стрессов. Основные направления лечения, применяемые в современной гастроэнтерологии для улучшения качества жизни человека:
- Диета. В рационе питания ограничивают потребление тугоплавких жиров и продуктов, содержащих консерванты. Полностью исключают употребление спиртных напитков. Показана витаминизированная диета с достаточной калорийностью.
- Препараты с желчегонным эффектом. Назначаются синтетические или растительные лекарственные средства, которые либо увеличивают концентрацию желчных кислот, либо повышают содержание водного компонента желчи. Специалисты отдают предпочтение мягким растительным препаратам.
- Санация очагов инфекции. Проводится выявление и лечение самых распространенных хронических источников инфекции — кариеса зубов и хронического тонзиллита. При обнаружении сопутствующей патологии желчевыводящих путей подбирается соответствующая патогенетическая терапия.
Прогноз и профилактика
Наличие болезни Дабина-Джонсона не влияет на продолжительность жизни и работоспособность, поэтому прогноз благоприятный. Синдром не прогрессирует. При соблюдении врачебных рекомендаций пациенты чувствуют себя хорошо. Больным необходимо воздерживаться от алкоголя и приема контрацептивов. Специфическая профилактика не разработана. Семьям с синдромом Дабина-Джонсона перед планированием беременности необходимо проконсультироваться с врачом-генетиком.
2. Клинические особенности течение синдрома Дабина-Джонсона у детей/ В.С. Березенко, М.Б. Диба, Ю.П. Резников// Современная педиатрия. — 2018.
4. Пигментные гепатозы: клинические особенности, пункционная биопсия, электронная микроскопия, диагноз, прогноз/ С.Д. Подымова// Практическая гастроэнтерология. — 2018.
Синдром Дубина—Джонсона и синдром Ротора: диагностика и лечение
Смежные специальности: гастроэнтеролог, терапевт.
Адрес: Санкт-Петербург, ул.Академика лебедева, д.4/2.
Синдром Дубина-Джонсона и синдром Ротора - это болезнь, которая наиболее распространена на Среднем Востоке (частота 1:1300) и диагностируется, преимущественно, у представителей мужского пола. Чаще всего страдают нарушением люди молодого возраста (около 70% случаев), редко заболевание развивается после 50-и лет. Данный синдром не оказывает влияния на длительность жизни человека.
Причины патологического состояния
У синдрома Дубина-Джонсона и синдрома Ротора аутосомно-рецессивный тип наследования. Болезнь развивается по причине врожденного дефекта — мутирует ген, который детерминирует протеин, осуществляющий транспортировку органических кислот.
В результате этого нарушается гепатобилиарная транспортировка органических анионов и билирубина. В крови возрастает концентрация конъюгированного билирубина, в моче выявляется билирубинурия.
Если обнаружена высокая концентрация прямого билирубина (больше 0,3 мг/дл) в комплексе с повышенным общим сывороточным билирубином (больше 2,0 мг/дл) или, если уровень прямого билирубина по отношению к сывороточному билирубину больше на 15%, диагностируют конъюгированную гипербилирубинемию (конъюгационную желтуху).
Первым проявлением синдрома Дубина—Джонсона может стать желтуха в период беременности или на фоне приема гормональных препаратов, используемых для ее предотвращения.
При данном расстройстве печень меняет свой цвет: орган приобретает зеленовато-серый или коричнево-черный оттенок. Макроскопически на печени обнаруживается наличие темных пятен, которые появляются в результате нарушения секреции метаболитов, таких как тирозин, триптофан, фенилаланин. Структура печени не нарушается. Пигмент может быть выявлен и в селезенке.
Развитие заболевания сопровождается слабостью, тошнотой, нарушением аппетита. Затем присоединяются болезненные ощущения в животе, гепатомегалия. Пальпация печени вызывает слабую боль. Моча приобретает темный оттенок, кал — обесцвечен. Как правило, печень имеет нормальные размеры, в редких случаях орган может увеличиваться на 1-2 см. Показатели крови остаются в норме.
Подобные симптомы имеет еще одно наследственное заболевание — синдром Ротора. Однако при такой патологии отсутствует печеночная пигментация. В остальном же,оба заболевания протекают одинаково.
Выявляют заболевание с помощью следующих методов:
- осмотра кожи;
- общего анализа мочи и крови (отмечается увеличение показателей желчных пигментов);
- исследования ферментов крови (показатели умеренно повышены);
- пробы с применением фенобарбитала (под влиянием данного средства концентрация билирубина снижается);
- бромсульфалеиновой пробы (экскреторная функция печени нарушена);
- ультразвукового обследования внутренних органов (в определенных случаях печень слегка увеличена, изменения формы, размеров, толщины стенок желчных протоков и пузыря отсутствуют, селезенка часто увеличена, конкременты не обнаруживаются);
- рентгенологическое обследование желчевыводящих путей (наполнение желчного пузыря и путей контрастным веществом запаздывает или полностью отсутствует);
- лапароскопии (диагностируется черный оттенок печени);
- пункционной биопсии печени (в гепатоцитах выявляется характерный пигмент).
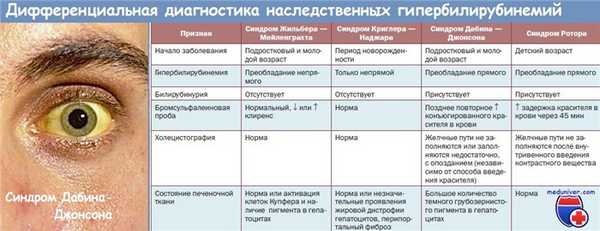
Следует дифференцировать синдром Дубина—Джонсона с синдромом Ротора, Жильбера, Криглера-Найяра, а также с таким заболеваниями, как механическая желтуха, гепатит, первичный билиарный цирроз. С этой целью выполняют бромсульфанеиновую пробу, проводят процедуру пункционной биопсии и лапароскопии.
Лечебные мероприятия

Специальной медикаментозной терапии данных наследственных патологий не существует. Лечение основано на следующих принципах:
- исключить факторы, провоцирующие обострение болезни (инфекционные заболевания, интенсивная физическая активность, стресс, прием спиртных напитков и гепатотоксичных препаратов).
- соблюдать специальную диету — ограничить употребление жирных и консервированных продуктов, в рацион включать пищу, богатую витаминами группы В.
- исключить инсоляцию.
- по назначению врача — принимать желчегонные препараты.
лечить имеющиеся заболевания желчевыводящих путей.
Прогноз заболевания положительный при условии соблюдения указанных рекомендаций в течение всей жизни.
Профилактические мероприятия
Если заболевание диагностировано у ребенка, то родителям необходимо проконсультироваться с квалифицированным врачом-генетиком при планировании последующего зачатия. Если родственники семейной пары страдают данной патологией, рекомендации аналогичны.
Синдромы Дубина-Джонсона, Ротора у новорожденного - клиника, диагностика
Синдром Дубина-Джонсона представляет собой еще один тип семейной желтухи. Это доброкачественное состояние, хотя печень ребенка с данным синдромом имеет сниженную способность секретировать некоторые органические анионы и контрастное вещество при проведении холецистографии.
Причина возникновения этого синдрома — мутация гена MDR2. У пациентов (обычно не в младенческом, а в более старшем возрасте) развивается конъюгированная гипербилирубинемия, не связанная с холестазом. Данный синдром — еще один пример дефекта канальцевого переносчика гепатоцита.
Поскольку экскреция желчных кислот не нарушена, термин «нехолестатическая желтуха» является в данном случае более точным; внепеченочные и внутрипеченочные желчные протоки всегда проходимы. С младенческого возраста могут отмечаться периодические эпизоды желтухи, спровоцированные в некоторых случаях инфекцией и неправильно истолкованные как острый гепатит из-за быстрого начала заболевания.
Результаты традиционных анализов, назначаемых для исследования функции печени, в норме, за исключением повышенного уровня общего билирубина (обычно менее 15 мг/дл) с преобладанием конъюгированного билирубина. Согласно данным морфологических исследований, печень имеет черный цвет из-за накопления меланиноподобного пигмента в лизосомах.
Синдром Ротора
Синдром Ротора похож на синдром Дубина-Джонсона, за исключением того, что при синдроме Ротора не отмечается пигментации гепатоцитов и секреция печенью контрастного вещества при проведении холецистографии — нормальная. Кроме того, первоначальный дефект у младенцев с синдромом Ротора заключается в дефиците внутриклеточных белков, связывающих анионы и служащих переносчиками их в клетку.
В качестве белка-переносчика может выступать глютатион-5-трансфераза. Дефицит этого внутриклеточного белка приводит к тому, что конъюгаты билирубина снова попадают в кровь вместо того, чтобы быть экскретированными после прохождения через канальцевую мембрану. При обоих описываемых синдромах отмечается повышение экскреции копропорфирина с мочой. Распознавание синдрома Ротора и синдрома Дубина-Джонсона представляется важным в плане предупреждения проведения ненужных дополнительных диагностических исследований.
По результатам гистологического исследования ткани печени можно предположить причину заболевания у ребенка, но безуспешные попытки специфической диагностики обусловили необходимость проведения такого исследования, которое помогло бы получить изображение «дерева» желчных протоков. Эту задачу решила гепатобилиарная сцинтиграфия.
У умерших детей с гепатоцеллюлярным холестазом при гистологическом исследовании печени было выявлено значительное количество аномалии, включая выраженное перипортальное воспаление и фиброз, а также диффузную трансформацию гигантских клеток. Примерно у 30% младенцев с гепатоцеллюлярным холестазом развивается прогрессирующая печеночная недостаточность, еще у 10%, переживших первые месяцы болезни, позднее выявляется хроническая патология, в т.ч. цирроз. Остальные 60% детей полностью выздоравливают.
В целом исходы для пациентов с этим заболеванием в настоящее время значительно улучшились благодаря успехам трансплантации печени.
Видео этиология, патогенез желтухи (повышения билирубина)
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: