Слабоумие плода. Монголизм эмбриона
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Болезнь Хантингтона (синдромХантингтона, хореяХантингтона или Гентингтона) — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 35-50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене IT-15. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Симптомы
Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это происходит в 35–44 года[22][23]. На ранних стадиях происходят небольшие изменения личности, когнитивных способностей и физических навыков[22]. Обычно первыми обнаруживают физические симптомы, так как когнитивные и психические расстройства не столь выражены в ранних стадиях[22]. Почти у всех пациентов болезнь Хантингтона в конечном итоге проявляется схожими физическими симптомами, но начало заболевания, прогрессирование и степень когнитивных и психических нарушений различаются у отдельных лиц[24][25].
Для начала заболевания наиболее характерна хорея — беспорядочные, неконтролируемые движения. Хорея в начале может проявляется в виде беспокойства, небольших непроизвольных или незавершённых движений, нарушения координации и замедления скачкообразных движений глаз[22].
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Чаще всего происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, может возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания.
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни[22]. Медицинская визуализация — такая, как компьютерная томография (КТ), магнитно-резонансная томография (МРТ) — показывает только видимую атрофию мозга на прогрессирующей стадии заболевания. Методы функциональной нейровизуализации — такие, как фМРТ и позитронно-эмиссионная томография (ПЭТ) — могут показать изменения в активности мозга до появления физических симптомов[4].
Для проведения генетической диагностики болезни Хантингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ аллеле[26]. Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако, отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Хантингтона[4].
Около 90% диагнозов болезни Хантингтона, основанных на обнаружении типичных симптомов и семейном анамнезе, подтверждаются генетическим тестированием. Большинство других расстройств с аналогичными симптомами называют ХГ-подобными расстройствами (англ. HD-like disorders, HDL)[28][29]. Причины большинства HDL заболеваний неизвестны. Известно лишь, что некоторые из них возникают в результате мутаций генов PRNP (HDL1), junctophilin 3 (HDL2), рецессивно наследуемого HTT гена (HDL3 — обнаружен у одной семьи и мало изучен) и гена, кодирующего ТАТА-связывающий белок (HDL4/SCA17)[29]. К другим заболеваниям с аутосомно-доминантным наследованием, которые схожи с болезнью Хантингтона, относят дентаторубро-паллидолюисовую атрофию и нейроферритинопатию[29].
Лечение
Химическая структура тетрабеназина, разрешённого для лечения болезни Хантингтона
Болезнь Хантингтона неизлечима, но существует лечение (купируется), способное облегчить некоторые симптомы.[30]
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Хантингтона[31], был утвержден в 2008 году в США[32]. Другие лекарства, помогающие уменьшить проявления хореи включают нейролептики и бензодиазепины[23]. Такие соединения как амантадин и ремацемид, находятся в стадии исследования, но показали положительные результаты[33]. Для облегчения таких симптомов как гипокинезия и ригидность мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии назначают вальпроевую кислоту[23].
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин, а при психозах и нарушениях поведения назначают атипичные антипсихотики[34].Хореический синдром чаще всего возникает вследствие ревматического энцефалита у детей (малая, или инфекционная, хорея) и при хорее Гентингтона. Хореический гиперкинез характеризуется беспорядочными быстрыми подергиваниями в разных мышцах, преимущественно в проксимальных отделах рук, мышцах лица (напоминают гримасничанье), иногда в мышцах живота и ног. Гиперкинезы могут быть односторонними или двусторонними. В поведении больного хореей отмечается суетливость, несоразмерность обычных двигательных актов. Все эти нарушения в двигательной сфере происходят на фоне выраженной мышечной гипертонии
47Наиболее распространенные наследственные заболевания, протекающие с нарушением психических функций.
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с дефектами в программном аппарате клеток, передаваемыми по наследству через гаметы. Термин употребляется в отношении полиэтиологических заболеваний, в отличие от более узкой группы — генные болезни.
Слабоумие - необратимое обеднение всей психической деятельности, сопровождаемое утратой или снижением полученных в прошлом знаний и навыков. Слабоумие бывает врожденным или возникает в результате перенесенных заболеваний.
ШИЗОФРЕНИЯ - психическая болезнь с тенденцией к хроническому течению. Причина заболевания неизвестна, нередко отмечается наследственная передача. По наследству передается не само заболевание, а схема обменных процессов заболевания. Не редко, когда передается по наследству нарушение обменных процессов головного мозга, человек может не заболеть этим заболеванием, т.е. это заболевание может не проявиться. Для проявления шизофрении, при наличие всех факторов, требуется "запускающий механизм". Это может быть алкоголь, наркотики, сильные стрессовые ситуации, неблагоприятные социальные условия и т.д.
Болезнь Гентингтона (БГ). Вызывается аутосомным доминантным геном. При наличии одного больного родителя риск передачи потомству составляет 50%. БГ – неврологическое расстройство , проявляющееся в любом возрасте . Хорея (быстрые, беспорядочные, непроизвольные, неконтролируемые движения лица и конечностей), раньше называвшаяся “пляской святого Витта”, встречается в семьях с нервным темпераментом.
Синдром Дауна (монголизм). Вызван лишней аутосомной 21-й хромосомой. У больного ребенка замедляется психофизическое развитие . Синдром встречается во всех расах и национальностях. Амниоцентез может выявить лишнюю хромосому в околоплодных водах матери
Болезнь Тэя — Закса (ранняя детская идиотия амавротическая) — редкое наследственное заболевание нервной системы.Болезнь проявляется в сильном нарушении моторных актов, восприятия и интеллектуальной деятельности. Для неё характерно наличие темно-красного пятна на сетчатке. Чаще всего проявляется в младенчестве. В таком случае, примерно с шести месяцев ребёнок становится менее активным, постепенно теряет зрение и слух. Заканчивается летальным исходом в возрасте 5 — 6 лет.
Синдром Туре́тта (болезнь Туретта, синдром Жиль де ла Туретта) — генетически обусловленное расстройство центральной нервной системы с манифестацией в детском возрасте, характеризующееся множественными моторными тиками и как минимум одним вокальным тиком.
48Участие психолога в их квалификации и преодолении имеющихся нарушений психической деятельности.
Методы работы психолога:
ñ Методы психогенетического анализа — генеалогический метод, метод анализа близнецовых пар, метод усыновления.
ñ Нарушения, связанные с изменением строения и числа хромосом (склонность к патологическим зависимостям, стерильность, склонность к асоциальному поведению, снижение интеллекта, синдром Тернера, синдром Клейнфельтера и др.).
ñ Проявление хромосомных нарушений во внешнем облике ребенка.
ñ Проективная диагностика нарушений, связанных с аутосомными мутациями (глухонемота, шизофрения, МДП, дебильность). Особенности наследования этих нарушений.
ñ Диагностика нарушений, вызванных мутациями, сцепленными с полом (гемофилия, дальтонизм). Особенности наследования этих нарушений.
ñ Подбор родителей при усыновлении детей с генетической отягощенностью.
ñ Разработка оптимальных психолого-педагогических маршрутов для детей с генетической отягощенностью.
Формы работы:
мини-лекции, дискуссии, решение задач по психогенетике, разбор сложных случаев из практики.
49Болезнь Дауна. Клинические проявления.
Синдро́м Да́уна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия, см. также плоидность). Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4 % случаев, и мозаичный вариант синдрома — 5 %.
лово «синдром» означает набор признаков или характерных черт. При употреблении этого термина предпочтительнее форма «синдром Дауна», а не «болезнь Дауна».[1]
При патоморфологическом исследовании выявляют аномалии нервной системы: недоразвитие мозга в целом или его отдельных частей, неправильное строение мозговых извилин, микроцефалию, гидроцефалию, нарушение дифференциации нервных клеток, их неправильное расположение, нарушение процессов миелинизации проводящих путей, патологию сосудистой системы мозга.
Нередки пороки сердца, крупных магистральных сосудов, других внутренних органов.
Заболевание можно диагностировать уже в период новорожденности на основании характерного внешнего вида ребенка: череп округлой формы, затылок скошен, косой разрез глаз, широка» переносица, добавочное веко, пятна светло-серого цвета на радужной оболочке, «готическое» нёбо, маленькие уши. Рот обычно полуоткрыт; язык толстый, часто высовывается изо рта, покрыт поперечными бороздами. Кисть широкая, пальцы кистей и стоп укорочены, мизинец часто искривлен
Иногда пальцы сросшиеся (частичная или полная синдактилия). На ладони нередко обнаруживается полная поперечная (обезьянья) складка.
У детей более старшего возраста фигура приземистая, конечности короткие, плечи опущены, голова наклонена вперед. Часты нарушения прикуса и аномалии зубов. Губы, особенно нижняя; утолщены.
Нижняя губа нередко вывернута. С большим постоянством выявляются значительные офтальмологические отклонения (близорукость, дальнозоркость, астигматизм).
У многих больных имеются эндокринные нарушения: ожирение, снижение основного обмена.
По-видимому, это связано с понижением функции щитовидной железы. К числу частых нарушений относятся сухость кожи, изменения слизистых оболочек, ногтей, волос вплоть до их полного выпадения.
В неврологическом статусе у больных могут быть выявлены сходящееся косоглазие, нистагм, асимметрия лица, легкие координаторные нарушения. Мышечный тонус обычно снижен.
Двигательные навыки развиваются с задержкой, отмечается неловкость движений.
Характерным признаком является недоразвитие интеллекта, степень этого недоразвития может варьировать. В большинстве случаев интеллект снижен до уровня имбецильности, реже до степени дебильности или идиотии.
Эмоции этих детей относительно сохранны, чего не наблюдается при других формах слабоумия. Дети с болезнью Дауна ласковы и очень привязаны к близким. Однако у них нередко отмечается неустойчивость настроения: повышенное настроение может сопровождаться аффективными вспышками или сменяться негативизмом. Дети с болезнью Дауна послушны и обладают хорошей способностью к подражанию, поэтому им легко привить навыки самообслуживания, их не трудно научить выполнять домашнюю работу или какие-либо другие простые трудовые операции.
Слабоумие плода. Монголизм эмбриона
Слабоумие плода. Монголизм эмбриона
Наиболее серьезные дефекты нервной системы внешне незаметны и не поддаются исправлению. Они проявляются в недостаточном умственном развитии человека, в его неспособности к разумному поведению и мышлению. В основе этого явления лежит непреложный закон «подобный рождает подобное».
Известно, что морфологическое строение мозга при слабоумии не отличается от строения мозга у заведомо нормального человека. Поэтому вопрос о происхождении слабоумия является скорее проблемой генетики, а не эмбриологии.
Можно, однако, надеяться, что когда мы будем больше знать о механизме, регулирующем эмбриональное развитие, то мы сможем обнаружить по крайней мере отдельные случаи, при которых слабоумие вызывается поправимыми факторами развития или внешней среды, а не суровыми законами наследственности, проявляющимися после слияния гамет.

Монголизм эмбриона
Морфологически более заметным, чем слабоумие, является состояние, известное как монголоидный идиотизм, или монголизм. Название происходит от косого положения глаз — одного из наиболее заметных внешних признаков этого состояния. Мы не знаем причин, вызывающих монголоидный идиотизм. Однако возможно, что при монголизме происходит нарушение многих процессов развития. Голова при этом состоянии маленькая и круглая вследствие того, что переднезадний диаметр головы ненормально укорочен. Язык увеличен и рот открыт, что придает лицу характерное выражение.
Подобно голове, головной мозг маленький, извилины полушарий менее сложны и более мелкие, чем это наблюдается в норме. Слабое развитие мозга указывает на его слабую функциональную способность. При монголоидном идиотизме умственное развитие больного соответствует таковому у детей 4—7 лет. Кровообращение у монголоидных идиотов недостаточное. Врожденные дефекты сердца и других внутренних органов встречаются у них особенно часто. Вполне возможно, что недостаточное кровообращение отчасти может быть причиной некоторых, как правило, сопутствующих монголизму состояний, например позднего прорезания молочных зубов.
Возможно также, что недостаточность кровообращения в большей степени, чем это обычно предполагают, обусловливает многие обнаруживающиеся при монголизме дефекты развития. Принимая во внимание обширные нарушения процессов развития, имеющие место у монголоидных идиотов, не следует удивляться, что они в большинстве случаев умирают очень рано — пятилетнего возраста достигают менее 60%.
Специфические дефекты развития головного мозга далеко не всегда сопровождаются какими-либо внешними аномалиями. Распознать их существование у человека можно только по его поведению. Например, может встречаться недостаточное развитие коры мозга. Структурно это проявляется в уменьшении поверхности коры и глубины борозд. На степень выраженности дефекта указывает также полнота обрастания корой островка (insula). Функционально дефекты этого типа выражаются значительным слабоумием.
Другим местным дефектом является частичное или полное недоразвитие мозолистого тела (corpus callosum). Удивительно, что этот серьезный морфологический дефект не вызывает существенного функционального нарушения, которое можно было бы заметить при жизни. Встречается также частичное или полное недоразвитие мозжечка. Степень этого уродства значительно варьирует. По-видимому, когда нарушение возникает относительно рано в ходе развития, то поражаются червячок (vermis) и полушария мозжечка. Если нарушение произойдет позднее, то в этом случае могут быть затронуты только одни полушария. У индивидуумов с такими дефектами не происходит утраты какой-либо определенной функции, а наблюдается лишь понижение или полное отсутствие координации в движениях.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром дауна (монголизм)

Врач акушер-гинеколог высшей категории, кандидат медицинских наук, член Ассоциации гинекологов-эндоскопистов РФ, член Ассоциации эндометриоза, член Общества репродуктивной медицины и хирургии, член European Society of Gynecology
В числе хромосомных патологий особое место занимает синдром Дауна – одно из наиболее распространенных генетических нарушений у новорожденных. Его основная причина – случайная генетическая мутация, вследствие которой в 21 паре хромосом появляется третья лишняя хромосома. Частота явления – приблизительно 1 случай на 600-800 малышей. Случайная мутация накладывает свой отпечаток на внешность ребенка уже на этапе внутриутробного развития, что значительно облегчает диагностику синдрома Дауна методом УЗИ.
Основные причины возникновения
Современная медицина называет сразу две причины болезни:
- Возраст матери. Это основной фактор риска для синдрома Дауна. Чем старше беременная женщина, тем выше риск рождения ребенка со случайной генетической патологией. В возрасте 30-40 лет риск генетического сбоя составляет 1/1000, после 42 лет – 1/60. Основной фактор – старение яйцеклеток, которые закладываются еще в период внутриутробного развития девочки и постепенно утрачивают способность к образованию генетически здорового плода. Также имеет значение возраст отца – до или после 45лет, когда вероятность рождения малыша с синдромом Дауна резко возрастает.
- Наследственный фактор. Причиной развития синдрома могут стать близкородственные браки, наличие заболевания у одного из родных ребенка. Также значение имеет возраст бабушки, в котором она родила дочь. Чем он выше, тем больше риск рождения внука с синдромом.
Важно помнить: синдром Дауна признан специалистами всех стран мира как случайная генетическая мутация. Она не зависит от экологической обстановки, уровня радиации, наличия вредного производства и прочих посторонних факторов.
Характерные внешние и прочие симптомы
Люди, являющиеся носителями лишней хромосомы, имеют характерную внешность:
- плоская переносица;
- монголоидный разрез глаз, из-за которого патология имеет второе название «монголизм»;
- плоское лицо и затылок.
Также в числе особенностей– некоторое отставание в развитии и сниженный иммунитет, не позволяющий организму сопротивляться внешним инфекциям. Все перечисленное не является ограничивающим фактором. Сегодня для них разработаны специальные методики обучения с первых месяцев жизни. При условии активности родителей дети с симптомами синдромома Дауна могут получить среднее образование и профессию, стать полноценными членами общества и завести собственную семью.
Осложнения
В зависимости от сложности формы синдрома Дауна у пациента могут отмечаться:
- врожденные пороки сердца;
- частые инфекционные заболевания;
- лейкемия;
- раннее наступление болезни Альцгеймера;
- остановка дыхания во сне;
- ожирение и т.д.
Диагностика
Выявление генетической аномалии возможно на ранних сроках беременности:
- УЗ-скрининг в период 11-13 недель оценивает размер воротникового пространства и размеры носовой кости плода;
- одновременно проводится анализ крови с уточнением количества хорионического гормона и плазменного протеина;
- на более поздних сроках беременности осуществляется забор тканей плода на предмет их генетического исследования: амниоцентез, биопсия волокон хориона или кордоцентез.
Т.к. речь идет о генетическом сбое, лечение синдрома Дауна заключается лишь в наблюдении за состоянием здоровья пациента и корректировкой осложнений основного заболевания.
Прогноз для пациентов
Сегодня средняя продолжительность жизни с генетической патологией приближается к 55-60 лет, тогда как еще несколько десятков лет назад они доживали только до 25 лет из-за неблагоприятных условий жизни.
Вероятность рождения ребенка с генетической аномалией у человека с синдромом Дауна составляет около 35-50%. Кроме того, при формировании плода у беременной женщины с заболеванием у будущего малыша могут возникнуть другие генетические сбои.
При этом риск онкологических заболеваний у таких больных сведен к нулю. Кроме того, родители отмечают радушие и неизменно хорошее настроение таких детей, их ласку, отзывчивость, способность легко идти на контакт и не обижаться на окружающих.
Профилактика

Полностью исключить риск рождения ребенка с генетической патологией 21-й пары хромосом не представляется возможным. Однако в силах будущих родителей сделать все возможное, чтобы укрепить собственное репродуктивное здоровье и исключить хромосомный сбой:
- следить за своим здоровьем, своевременно обращаться за медицинской помощью для лечения выявленных заболеваний;
- вести здоровый и активный образ жизни, заниматься спортом, чтобы в яйцеклетки поступало достаточное количество кислорода;
- правильно питаться, обогащая рацион здоровыми продуктами с высоким содержанием витаминов и микроэлементов;
- поддерживать иммунную систему;
- следить за весом, т.к. его отклонение в любую сторону может стать причиной гормонального сбоя и нарушения процесса созревания половых клеток;
- своевременно проходить УЗ-обследование во время беременности, чтобы выявить у плода генетический сбой на первых неделях внутриутробного развития.
Наблюдение за пациентами с синдромом Дауна в АО «Медицина» (клиника академика Ройтберга) в Москве
В АО «Медицина» (клиника академика Ройтберга) функционирует Центр по работе с особыми детьми. Педиатры и профильные специалисты клиники в ЦАЛ Москвы готовы к работе с пациентами с синдромом Дауна независимо от их возраста и общего состояния организма.
Пациентам гарантировано внимательное и ответственное отношение, индивидуальный подход, конфиденциальность личных данных и достижение видимых результатов назначенного лечения или профилактики синдрома Дауна. Все необходимые диагностические мероприятия можно пройти в клинике, чтобы получить быстрые и достоверные результаты.
Частые вопросы о заболевании
Сколько живут люди с синдромом Дауна?
Средняя продолжительность жизни пациентов с синдромом Дауна составляет сегодня около 50 лет, увеличившись более чем в 2 раза относительно статистики середины прошлого столетия. На срок жизни влияет наличие осложнений в организме, вызванных генетическим сбоем, условия жизни, наследственность и т.д. При условии квалифицированного медицинского наблюдения продолжительность жизни пациентов удается продлить и сделать ее более комфортной.
Как определить заболевание во время беременности?
Уточнить риск развития плода с генетической аномалией можно с помощью ультразвукового исследования на ранних сроках беременности, а также при лабораторном исследовании крови на предмет содержания белков и гормонов в организме будущей матери. При наличии серьезных подозрений на генетический сбой на более поздних сроках исследуется биоматериал плода, полученный путем биопсии или забора околоплодной жидкости.
Лечится ли синдром Дауна?
Преимплантационный генетический скрининг (PGS) в клинической практике
В процессе старения нарушается процесс образования женских половых клеток и в 25% случаев мужских половых клеток. Возрастные процессы оказывают влияние на деление и созревание половых клеток, существует риск при созревании получить больше хромосом, чем нужно. И если эта «особенная» клетка примет участие в оплодотворении, образовавшийся зародыш будет иметь 23 пары хромосом и еще + 1, что скажется на его дальнейшем развитии.
Так например, больной синдромом Дауна организм насчитывает не две, а три хромосомы из 21 пары.
Риск заболеваний генетического рода у будущего ребенка увеличивается в случае, когда возраст матери превышает 35 лет, а отца превышает 45-летний возраст.
Синдром Дауна наиболее часто выражается в форме стандартной трисомии, когда во всех клетках организма полностью утраивается 21 хромосома. Эта форма заболевания занимает 94% всех случаев.
Около 4% случаев занимает форма транслокации, смещения 21 хромосомной пары на остальные хромосомы.
Самая редко встречающаяся форма заболевания - мозаичная (около 2% всех случаев). При ней утроенная 21 хромосома содержится только в некоторых клетках организма человека. Заболевший человек, при такой форме имеет нормальную внешность, развитый интеллект.
Мозаичный синдром Дауна при беременности определить весьма проблематично, так как большинство клеток плода будут наделены свойствами обычного кариотипа. Подростки с данной формой заболевания по внешнему виду могут иметь сходства с внешностью при диагнозе синдром Дауна, но, несмотря на это, отлично учиться в школе на уровне с ровесниками. Подтвердить диагноз при мозаичной форме синдрома Дауна достаточно сложно, так как лишь у 10% клеток из общего количества трисомная форма 21 хромосомы. Анализ крови на синдром Дауна подразумевает сдачу крови в больших количествах на кариотип — только в таком случае возможен точный диагноз.
Синдром Дауна в трисомной форме делает мужчин бесплодными, а мозаичная форма делает возможной функцию деторождения, но рожденные дети в 98% случаев будут наделены синдромом Дауна. К сожалению, с диагнозом синдром Дауна, при всех его формах, родить здоровое потомство практически невозможно.
Как найти выход с помощью PGS
Истории из жизни или глазами пациентов:
«Мой младший брат родился с синдромом Дауна»
Около тридцати я вышла замуж, и мы начали стараться завести ребенка. К сожалению, проходило время, а я не беременела. У меня никогда не было серьезных проблем со здоровьем, порой были нерегулярные менструации, но все гинекологи до сих пор утверждали, что я спокойно могу стать мамой. Я начала беспокоиться, когда в течение двух лет ничего не происходило. Наконец, я уговорила мужа, чтобы мы проверили, что не в порядке. В клинике нам пришлось пройти много обследований и ответить на ряд детальных вопросов. Слава богу, доктор, которая с нами беседовала, была очень приятной. Мы как-то сразу нашли общий язык, несмотря на то, что мой любимый в самом начале «лечения» держался отстраненно. После приема мы решили также обратиться за консультацией к генетику. Я знала, что порок моего брата может означать, что у меня есть какая-то семейная генетическая отягощенность. Это подтвердил и специалист. Сомнения, однако, остались, тем более, что нам не удавалось естественным путем зачать ребенка. Результаты обследований не принесли ничего нового. Идиопатическое бесплодие, то есть бесплодие с неизвестной причиной, – так звучал диагноз. Мы решили подождать еще несколько месяцев, а если терпения не хватит дожидаться чуда, тогда довериться врачам. Под конец года мы решились попробовать ЭКО. В клинике нам предложили «ЭКО без риска» – наши эмбрионы должны были быть обследованы с точки зрения генетической отягощенности, имеющейся в моей семье. Я думаю, что это было самым правильным решением, и я благодарна врачам за помощь. Оказалось, что из четырех эмбрионов, которые правильно развивались, два были больными. Не знаю, много это или мало, но, когда я смотрю на наших близнецов, как они развиваются, смеются, играют – я так счастлива, что они здоровы.
Я столкнулась с разными мнениями и на тему ЭКО, и относительно диагностики, которую мы прошли – но у меня на это лишь один ответ. Никто не поймет, что чувствует бесплодный человек, если сам этого не испытает… Я ни секунды не жалела, что мы стали лечиться, ни разу не подумала, что поступила неправильно. Совсем наоборот – я всей душой чувствую, что это того стоило.
Рождение здорового ребенка после преимплантационного генетического скрининга эмбрионов у мужчины с синдромом Дауна (Journal of Assisted Reproduction and Genetics; сентябрь 2015г)
В клинику репродуктивного здоровья (Alameda County Medical Center, Калифорния), обратилась пара с 6-летней историей первичного бесплодия: женщина 26 лет с нормальным хромосомным набором и 29-летний мужчина с синдромом Дауна (трисомия по 21 паре хромосом). В результате проведения контролируемой стимуляции овуляции было получено 33 яйцеклетки, 29 из которых оплодотворили методом ИКСИ. На 5 сутки развития 13 эмбрионов хорошего качества подверглись биопсии трофэктодермы для проведения предимплантационного генетического скрининга (ПГС), после чего были заморожены методом витрификации.
Анализ показал, что 12 из 13 (92%) имели нормальный хромосомный набор.
После переноса здорового эмбриона наступила беременность, в процессе которой был проведен пренатальный генетический скрининг, подтвердивший отсутствие патологии. В результате на 41 неделе с помощью операции кесарева сечения на свет появился здоровый мальчик.
Таким образом, преимплантационный генетический скрининг (PGS) – метод, который позволяет ограничить риск пороков и болезней у ребенка и дает многим парам шанс на счастливое родительство. Представляет собой исследование клеток эмбриона перед его подсадкой в рамках цикла ВРТ (вспомогательных репродуктивных технологий). В матку женщины вводятся исключительно те эмбрионы, у которых не выявлено хромосомных нарушений.
Благодаря полученным знаниям, можно свести к минимуму риск возникновения у плода серьезных (а часто смертельных) генетических пороков, а также других хромосомных аномалий, для которых характерны выраженная умственная отсталость и наличие физических пороков развития.
Выполнение диагностики PGS в рамках процедуры ЭКО – это, главным образом, психологический комфорт для пациентов, которые опасаются рождения больного ребенка или имеют показания к процедуре!
Мой маленький плод Революционное открытие победило синдром Дауна и другие патологии еще в утробе
Биологи обнаружили, что у зародышей есть система безопасности против дефектных клеток с генетическими нарушениями. Даже если такие клетки составляют добрую половину эмбриона, организм сможет избавиться от них и вполне нормально развиваться. Чтобы понять это, ученым пришлось создать химерный зародыш из здоровых и больных клеток. «Лента.ру» ознакомилась с исследованием и выяснила интересные детали.
Если на ранних стадиях развития эмбриона образуются аномальные клетки, это необязательно признак того, что ребенок родится с врожденными пороками. Новое исследование Кембриджского университета раскрывает механизмы, предотвращающие нарушения в развитии организма. Оказывается, аномальные клетки уничтожаются и заменяются здоровыми.
Группа исследователей с кафедры физиологии и нейробиологии изучала эмбрионы мышей, в которых некоторые клетки содержали ненормальное число хромосом. Как правило, в каждой клетке человеческого эмбриона 23 пары хромосом. 22 — аутосомы, парные хромосомы, одинаковые для мужского и женского организма. Одна пара — это половые хромосомы, отличающиеся у мужчин (XY), но одинаковы у женщин (XX). При анеуплоидии возникают изменения в числе хромосом. Например, от пары остается одна хромосома или, наоборот, появляется третья лишняя. Ситуация, когда вместо двух хромосом — три копии одной хромосомы, называется трисомией. Возможны также две (тетрасомия) и три добавочные хромосомы (пентасомия). Анеуплоидия приводит к расстройствам в развитии человека. Самый известный пример — синдром Дауна, при котором у двадцать первой хромосомы три копии.
Синдром Дауна является единственной жизнеспособной трисомией. Другие трисомии, например синдром Патау (хромосома 13) и синдром Эдвардса (хромосома 18), вызывают серьезные нарушения развития и раннюю смертность после рождения. Другие виды трисомий в аутосомах приводят к гибели эмбриона, при этом самой часто встречающейся аномалией является трисомия по 16-й хромосоме, которая приводит к выкидышу. Моносомии вызывают более тяжелые последствия, чем трисомии, и все являются летальными для зародышей. Единственное исключение — синдром Шерешевского-Тернера, который встречается у женщин и вызывается утратой одной из половых хромосом. Эта болезнь сопровождается нарушениями физического и психического развития, а также карликовостью. В противоположность этому дополнительные половые хромосомы оказывают мягкое влияние на развитие человеческого организма, однако могут негативно сказываться на умственном развитии.
Беременным женщинам, особенно в возрасте, поскольку их дети наиболее подвержены риску анеуплоидии, предлагают тесты, позволяющие предсказать вероятность генетических аномалий. Между 11-й и 14-й неделями беременности будущей матери могут провести биопсию хориона. Врач извлекает кусочки ткани плаценты, и клетки анализируются на количество хромосом. При другом тесте — амниоцентезе — изучаются клетки из амниотической жидкости (околоплодных вод). Этот тест проводится в течение 15-20 недель беременности, и его результаты более точны.
Внимание авторов нового исследования привлек один случай. Биопсия хориона одной из беременных женщин показала, что около четверти клеток плаценты были с генетическими аномалиями, однако ребенок родился здоровым. Ученые задумались о причине возникновения аномальных клеток в тканях, окружающих эмбрион и о том, в какой степени по ним можно судить о риске патологии.
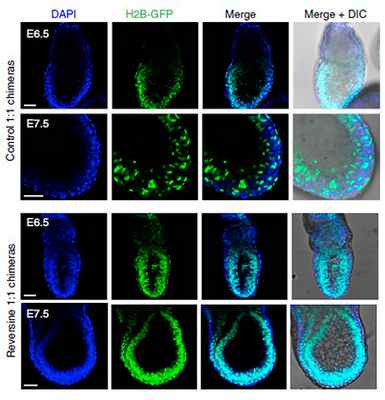
Изображение химерного эмбриона с обычными и аномальными клетками
Изображение: Helen Bolton / Gurdon Institute, University of Cambridge
Аномальные клетки с нарушениями в структуре и количестве хромосом наблюдаются примерно в 80-90 процентах человеческих эмбрионов в предымплантационном периоде беременности, когда оплодотворенная яйцеклетка движется по маточной трубе к матке. Эмбрион содержит как нормальные, так и нездоровые клетки. Это происходит из-за неизбежных ошибок в митозе во время дробления — раннего этапа эмбрионального развития, когда яйцеклетка разделяется на более мелкие клетки или бластомеры. Если в тканях эмбриона находятся генетически различающиеся клетки, говорят о хромосомном мозаицизме. Считается, что именно мозаицизм — основная причина неудачных беременностей при искусственном оплодотворении. Однако, хотя хромосомный мозаицизм очень часто встречается в ранних эмбрионах, на более поздних стадиях он не так выражен.
Как у мышей, так и у человека предымплантационное развитие завершается образованием бластоцисты — шара, состоящего из десятков или сотен клеток. В состав бластоцисты входит несколько групп клеток — трофобласт и примитивная эндодерма, которые формируют плаценту и желточный мешок. Кроме того, существует третья группа клеток, составляющую эмбриональный эпибласт, который в дальнейшем становится плодом. Развитие всех этих зародышевых клеток можно разделить на два этапа. На первом этапе клетки наружной части эмбриона формируют трофобласт, в то время как внутренние клетки образуют эмбриобласт. На втором этапе эмбриобласт дает начало эмбриональному эпибласту и примитивной эндодермы. Правильное развитие групп клеток на ранних стадиях необходимо для всего последующего эмбриогенеза.
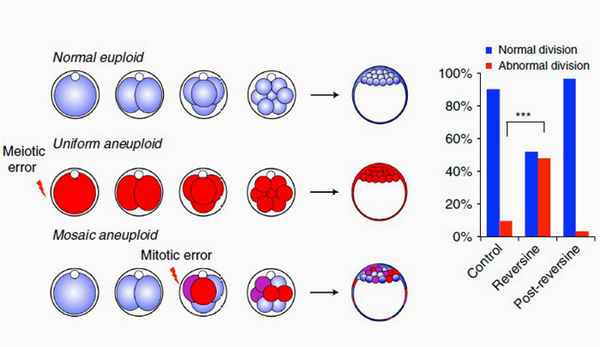
Эмбрионы с мозаичной анеуплоидией содержат здоровые клетки и клетки с различными генетическими аномалиями
Изображение: Helen Bolton / Gurdon Institute, University of Cambridge
Обработка эмбрионов реверсином уменьшала количество клеток в каждой из групп, хотя все группы продолжали правильно развиваться и морфология эмбриона оставалась ненарушенной. Однако на поздних стадиях зародыш погибал. Это напоминает судьбу эмбрионов, в чьих клетках отсутствуют гены, участвующие в синтезе кинетохоров — белковых структур на хромосоме, к которым крепятся веретена деления. Для таких эмбрионов характерна мозаичная анеуплоидия, и они гибнут на поздних стадиях развития.
Ученые визуализировали развитие зародышевых химер с помощью замедленной съемки высокого разрешения, позволяющей разглядеть каждую клетку в эмбрионе. Результаты показали, что у эмбрионов, где здоровых и аномальных клеток было поровну, клетки с генетическими нарушениями уничтожались в процессе апоптоза — программируемой гибели клеток, хотя плацентарные клетки сохраняли жизнеспособность. Это позволило нормальным клеткам одержать верх, и все клетки эмбриона оказались здоровыми. В случае, когда на одну здоровую клетку приходилось три аномальных, клетки с нарушениями выжили, однако доля нормальных увеличивалась.
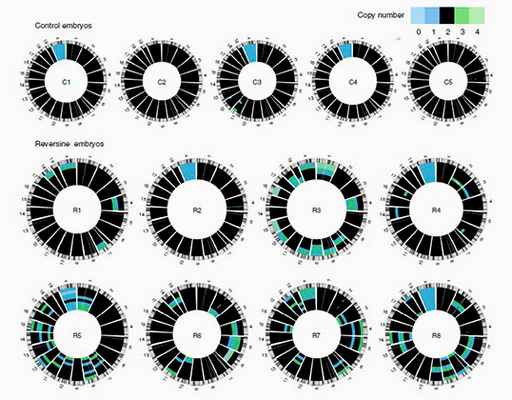
Анеуплоидные вариации в хромосомах различных эмбрионов, обработанных реверсином
Изображение: Helen Bolton / Gurdon Institute, University of Cambridge
Механизмы, активирующие гибель клеток, определить пока не удалось. Однако проведенное исследование — первое, непосредственно демонстрирующее постепенное исчезновение аномальных клеток из тканей эмбриона на ранних стадиях развития. Также впервые были получены доказательства гипотезы о том, что за гибель клеток с генетическими нарушениями отвечает апоптоз. Интересно то, что когда химерные зародыши с половиной дефектных клеток трансплантировались в матку самок мышей, степень выживаемости эмбрионов оставалась такой же высокой, как и в норме.
Эти результаты имеют большое значение для медицины, в частности для биопсии эмбриональных тканей. Теперь понятно, почему зародыш выживает, хотя анализы тканей плаценты дают плохой прогноз. Также показано, что более надежная биопсия клеток из самой бластоцисты может быть безопасной и ничем не вредит эмбриону.
Читайте также: