Сочетание нефронофтиза и аутосомной доминантной тубулоинтерстициальной болезни почек
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Хронический тубулоинтерстициальный нефрит у детей. Диагностика и лечение
У детей это состояние чаще всего развивается при врожденной почечной патологии, например обструкции мочевых путей или пузырно-мочеточниковом рефлюксе. Хронический тубулоинтерстициальный нефрит может быть идиопатическим, хотя это более характерно для взрослых. Аналогичные гистологические изменения наблюдаются при наследственных заболеваниях, называемых комплексом ювенильный нефронофтиз/медуллярная кистозная болезнь. Ювенильный нефронофтиз наследуется как аутосомно-рецессивный признак.
В нашей стране он встречается редко, но в странах Европы обусловливает 10-20 % случаев терминальной стадии ХПН. У больных, как правило, имеет место полиурия, задержка роста, необъяснимая анемия; ХПН развивается в позднем детском или подростковом возрасте. К вариантам ювенильного нефронофтиза с внепочечными проявлениями относят синдром Сениора-Локена (пигментная дегенерация сетчатки), синдром Жобера и окуломоторную апраксию типа Когана.
Медуллярный кистозный комплекс — аутосомно-доминантное заболевание, проявляющееся только в зрелом возрасте. Преимущественно у девочек-подростков встречается редкий аутоиммунный синдром тубулоинтерстициальный нефрит с у увеитом, при котором, кроме хронического тубулоинтерстициального нефрита и иридоциклита, обнаруживаются костномозговые гранулемы. Наконец, хронический тубулоинтерстициальный нефрит наблюдается при любых формах прогрессирующей болезни почек, независимо от ее причины, и наиболее важным предиктором развития терминальной стадии почечной недостаточности является тяжесть поражения интерстициальной ткани.
Патогенез и патоморфология тубулоинтерстициального нефрита у детей. Патогенез хронической формы этой патологии, как и острой, остается неясным, но имеющиеся данные свидетельствуют о роли иммунных механизмов. Макроскопически почки бледные и уменьшение. При микроскопическом исследовании обнаруживается атрофия канальцев и фиброз интерстициальной ткани с очагами лимфоцитарного воспаления. При ювенильном нефронофтизе часто находят мелкие кисты на границе коркового и мозгового вещества почек.
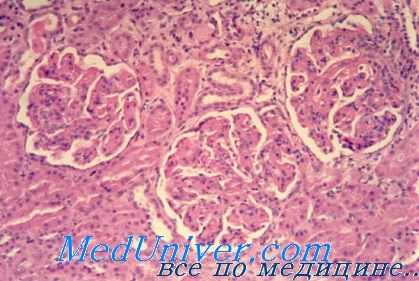
При первичном хроническом тубулоинтерстициальном нефрите до поздних стадий болезни клубочки остаются относительно сохранными. Когда тубулоинтерстициальный нефрит является следствием поражения клубочков, имеются признаки первичного заболевания.
Клинические проявления тубулоинтерстициального нефрита у детей. Клинические проявления хронического тубулоинтерстициального нефрита часто неспецифичны и просто отражают ХПН. Характерны задержка роста, полиурия, полидипсия и недержание мочи. Часто наблюдается анемия, особенно при ювенильном нефронофтизе. Поражение канальцев обычно сопровождается потерей соли, поэтому высокое АД отмечается редко.
Диагностика тубулоинтерстициального нефрита у детей. На хронический тубулоинтерстициальный нефрит указывают признаки и симптомы повреждения почечных канальцев, такие как полиурия и повышение уровня креатинина в сыворотке в сочетании с анамнестическими данными, свидетельствующие о хроническом течении болезни (длительный период недержания мочи или анемия, резистентная к препаратам железа).
УЗИ позволяет получить дополнительные данные о хронической природе заболевания (маленькие почки с повышенной эхогенностью, кисты в корково-мозговом веществе, характерные для ювенильного нефронофтиза, или признаки обструкции мочевых путей). При цистоуретрографии можно обнаружить пузырно-мочеточниковый рефлюкс или аномалии мочевого пузыря. При неясной причине болезни не исключена биопсия почек. Однако на поздних стадиях она не имеет диагностического значения, так как в терминальной стадии ХПН гистологические изменения (фиброз и воспаление канальцев) одинаковы при различных заболеваниях.
Лечение и прогноз тубулоинтерстициального нефрита у детей. Лечение направлено на поддержание водно-электролитного обмена и устранение нефротоксичных агентов. При обструктивной уропатии могут потребоваться солевые добавки и применение связывающей калий смолы. Профилактика инфекций с помощью антибиотиков в этих случаях замедляет развитие почечной патологии. Прогноз при хроническом тубулоинтерстициальном нефрите в значительной степени зависит от характера основного заболевания.
У больных с обструкцией мочевых путей или пузырно-мочеточниковым рефлюксом степень повреждения канальцев может быть разной; соответственно и болезнь имеет различное течение: ХПН может достичь терминальной стадии как через месяцы, так и через годы. При ювенильном нефронофтизе эта стадия всегда развивается уже к подростковому возрасту.
Интерстициальный нефрит ( Тубулоинтерстициальный нефрит )
Интерстициальный нефрит — это острое или хроническое негнойное воспаление стромы и канальцев почек, обусловленное гиперергической иммунной реакцией. Проявляется болями в пояснице, нарушением диуреза (олигоанурией, полиурией), интоксикационным синдромом. Диагностируется с помощью общего и биохимического анализов мочи, крови, УЗИ почек, определения β2-микроглобулина, гистологического исследования биоптата. Схема лечения сочетает детоксикацию при отравлениях, этиопатогенетическую терапию основного заболевания с назначением иммуносупрессоров, антигистаминных средств, антикоагулянтов, антиагрегантов. В тяжелых случаях требуется проведение ЗПТ, трансплантация почки.
МКБ-10
Общие сведения
Особенностью интерстициального нефрита является вовлечение в патологический процесс межуточной ткани, тубулярных структур, кровеносных и лимфатических сосудов без распространения на чашечно-лоханочную систему и грубой гнойной деструкции органа. Поскольку ведущую роль в патогенезе расстройства играет тубулярная дисфункция, в настоящее время заболевание принято называть тубулоинтерстициальным нефритом (ТИН).
По данным масштабных популяционных исследований, острые варианты интерстициального воспаления составляют до 15-25% всех случаев острого повреждения почек. Распространенность хронических форм заболевания по результатам пункционной нефробиопсии колеблется от 1,8 до 12%. Патология может развиться в любом возрасте, однако чаще наблюдется у 20-50-летних пациентов.
Причины
Поражение ренального канальцевого аппарата и межуточной ткани имеет полиэтиологическую основу, при этом роль отдельных повреждающих факторов отличается в зависимости от характера течения процесса. Основными причинами острого негнойного интерстициального воспаления почек, по наблюдениям специалистов в сфере урологии и нефрологии, являются:
- Прием нефротоксичных препаратов. Более 75% случаев острого тубулоинтерстициального нефрита развивается у чувствительных пациентов после приема антибиотиков, сульфаниламидов, НПВС, реже — тиазидных диуретиков, противовирусных средств, анальгетиков, барбитуратов, иммунодепрессантов.
- Аналогичный эффект могут вызывать вакцины и сыворотки.
- Системные процессы. У 10-15% больных патологические изменения в интерстициальной ткани и канальцах ассоциированы с аутоиммунными болезнями (синдромом Шегрена, СКВ), саркоидозом, лимфопролиферативными заболеваниями. В эту группу причин также входят случаи метаболических нарушений (гиперурикемии, оксалатемии) и острых токсических нефропатий.
- Инфекционные агенты. Хотя воспаление носит негнойный характер, у 5-10% пациентов оно возникает на фоне или после перенесенного инфекционного процесса. Интерстициальные формы нефрита могут осложнить течение бруцеллеза, иерсиниоза, цитомегаловирусной инфекции, лептоспироза, риккетсиоза, шистоматоза, токсоплазмоза, других инфекций, сепсиса.
- Неустановленные факторы. До 10% случаев внезапно развившегося нефрита с поражением интерстиция, канальцев имеют неуточненную этиологию и считаются идиопатическими. У части пациентов острая почечная патология сочетается с клиникой воспаления сосудистой оболочки глаз (синдром тубулоинтерстициального нефрита с увеитом).
Как и острые формы заболевания, хронический тубулоинтерстициальный нефрит может сформироваться на фоне длительного приема нефротоксичных лекарственных средств (в первую очередь НПВС, цитостатиков, солей лития), отравления ядами (солями кадмия, свинца). Патология часто возникает у пациентов с метаболическими расстройствами (подагрой, цистинозом, повышенными уровнями оксалатов, кальция в крови), аллергическими и аутоиммунными болезнями.
Хронический ТИН осложняет течение туберкулеза, заболеваний крови (серповидно-клеточной анемии, синдрома отложения легких цепей). У больных с аутосомно-доминантной тубулоинтерстициальной болезнью негнойный нефрит имеет наследственную основу. При длительной постренальной обструкции мочевыводящих путей (везикоуретеральном рефлюксе, аденоме предстательной железы, мочеточниково-влагалищных свищах и т. п.), атеросклерозе ренальной артерии, гломерулопатиях хроническое интерстициальное воспаление является вторичным.
Патогенез
Механизм развития интерстициального нефрита зависит от характера, интенсивности действия повреждающего фактора. Зачастую воспаление имеет аутоиммунную основу и провоцируется осаждением циркулирующих в крови иммунных комплексов (при лимфопролиферативных процессах, системной красной волчанке, приеме нестероидных противовоспалительных средств) или антител к базальной мембране канальцев (при интоксикации антибиотиками, отторжении трансплантата).
При хронизации процесса важную роль играет патологическая активация макрофагов и T-лимфоцитов, вызывающих протеолиз тубулярных базальных мембран и усиливающих перекисное окисление с образованием свободных радикалов. Иногда канальцевый эпителий повреждается в результате селективной кумуляции и прямого разрушающего действия нефротоксичного вещества, реабсорбируемого из первичной мочи.
Локальное выделение медиаторов воспаления в ответ на действие повреждающего фактора вызывает отек интерстиция и спазм сосудов, который усугубляется их механическим сдавлением. Возникающая ишемия почечной ткани потенцирует дистрофические изменения в клетках, снижает их функциональность, в части случаев провоцирует развитие папиллярного некроза и массивную гематурию.
Из-за повышения давления в канальцах и снижения эффективного плазмотока вторично нарушается фильтрующая способность гломерулярного аппарата, что приводит к почечной недостаточности и увеличению уровня сывороточного креатинина. На фоне отека межуточной ткани и повреждения канальцевого эпителия снижается реабсорбция воды, усиливается мочевыделение.
При остром течении нефрита постепенное уменьшение отека интерстициального вещества сопровождается восстановлением ренального плазмотока, нормализацией скорости клубочковой фильтрации и эффективности канальцевой реабсорбции. Длительное присутствие повреждающих агентов в сочетании со стойкой ишемией стромы на фоне нарушений кровотока влечет за собой необратимые изменения эпителия и замещение функциональной ткани соединительнотканными волокнами.
Склеротические процессы усиливаются за счет стимуляции пролиферации фибробластов и коллагеногенеза активированными лимфоцитами. Существенную роль в возникновении гиперергической воспалительной реакции играет наследственная предрасположенность.
Классификация
При систематизации клинических форм интерстициального нефрита принимают в расчет такие факторы, как наличие предшествующей патологии, остроту возникновения симптоматики, развернутость клинической картины. Если острое межуточное воспаление развивается у ранее здоровых пациентов с интактными почками, процесс считается первичным. При вторичном тубулоинтерстициальном нефрите почечная патология осложняет течение подагры, сахарного диабета, лейкемии и других хронических болезней. Для прогнозирования исхода заболевания и выбора оптимальной терапевтической тактики важно учитывать характер течения воспалительного процесса. Урологи и нефрологи различают две формы интерстициального воспаления:
- Острый нефрит. Возникает внезапно. Сопровождается значительными морфологическими изменениями стромы, канальцев, зачастую – обратимыми. Гломерулы обычно не повреждаются. Протекает бурно с выраженной клинической симптоматикой тубулярного поражения и вторичного нарушения клубочковой фильтрации. Часто наблюдается быстрое двухстороннее снижение или полное прекращение функции почек. Острые формы межуточного нефрита служат причиной 10-25% острой почечной недостаточности. Несмотря на серьезный прогноз, своевременное назначение адекватной терапии позволяет восстановить функциональные возможности органа.
- Хронический нефрит. Морфологические изменения развиваются постепенно, преобладают процессы фиброзирования интерстициальной ткани, атрофии канальцевого аппарата с его замещением соединительной тканью и исходом в нефросклероз. Возможна вторичная гломерулопатия. Симптоматика нарастает медленно, при выраженных склеротических процессах является необратимой. У 20-40% пациентов с хронической почечной недостаточностью нарушение фильтрующей функции почек вызвано именно тубулоинтерстициальным нефритом. Прогноз заболевания серьезный, при возникновении ХПН необходимо проведение ЗПТ и пересадка почки.
При остром воспалении оправдано выделение нескольких вариантов заболевания с разной выраженностью симптомов. Для развернутой формы нефрита характерна классическая клиническая картина. Отличительной особенностью тяжелого воспаления является ОПН с анурией, требующая срочного проведения заместительной почечной терапии. При благоприятно протекающем абортивном воспалении отсутствует олигоанурия, преобладает полиурия, концентрационная функция восстанавливается за 1,5-2 месяца. При развитии интерстициального очагового нефрита симптоматика стертая, превалирует нарушение реабсорбции мочи.
Симптомы интерстициального нефрита
Признаки заболевания неспецифичны, сходны с проявлениями других видов нефрологической патологии. Клиника зависит от особенностей развития воспалительного процесса. При остром нефрите и обострении хронического воспаления наблюдаются нарушения общего состояния — головная боль, ознобы, лихорадка до 39-40° С, нарастающая слабость, утомляемость. Возможно повышение артериального давления. В моче появляется кровь.
Пациент жалуется на сильные боли в пояснице, количество мочи резко уменьшается вплоть до анурии, которая впоследствии сменяется полиурией. При прогредиентном заболевании больного беспокоят тупые боли в области поясницы, незначительное снижение объема суточной мочи, папулезная сыпь. Иногда наблюдается субфебрилитет. О возможном снижении фильтрационной способности органа при хроническом варианте нефрита свидетельствует появление симптомов уремической интоксикации — тошноты, рвоты, кожного зуда, сонливости.
Осложнения
При отсутствии адекватной терапии острый интерстициальный нефрит зачастую переходит в хроническую форму. Изменения в почечном интерстиции со временем приводят к снижению количества функционирующих нефронов. Следствием этого является развитие хронической почечной недостаточности, инвалидизирующей пациента и требующей проведения заместительной терапии. Воспалительный процесс может вызвать активацию ренин-ангиотензин-альдостероновой системы, стимулировать повышенный синтез вазоконстрикторных веществ, что проявляется стойкой артериальной гипертензией, рефрактерной к медикаментозной терапии. Нарушение синтеза эритропоэтинов при хронических нефритах интерстициального типа становится причиной тяжелых анемий.
Диагностика
В связи с неспецифичностью клинической симптоматики при постановке диагноза интерстициального нефрита важно исключить другие причины острой или хронической нефропатии. Как правило, окончательная диагностика заболевания проводится на основании результатов гистологического исследования с учетом вероятных повреждающих факторов. Рекомендованными методами лабораторно-инструментального обследования являются:
- Общий анализ мочи. Характерна протеинурия — от небольшой и умеренной (суточное выделение с мочой 0,5-2 г белка) до нефротической (более 3,5 г белка /сут). У большинства больных определяется эритроцитурия, лейкоцитурия с наличием эозинофилов и лимфоцитов в моче. Возможна цилиндрурия. В анализе отсутствуют бактерии. Плотность мочи зависит от формы и стадии нефрита.
- УЗИ почек. Для острого интерстициального процесса типичны нормальные или несколько увеличенные размеры почек, повышение кортикальной эхогенности. При хроническом нефрите органы уменьшены, эхогенность усилена, у некоторых пациентов отмечается деформация контура. Исследование дополняют УЗДГ почек, выявляющим нарушение ренальной гемодинамики.
- Биохимический анализ крови. Результаты показательны при возникновении почечной недостаточности. Характерными признаками нарушения гломерулярной фильтрации служат повышение сывороточных уровней креатинина, мочевой кислоты, азота. Соответствующие изменения выявляются при проведении нефрологического комплекса и подтверждаются пробой Реберга.
- Бета-2-микроглобулин. Специфическим маркером нарушения реабсорбции в тубулярном аппарате является повышение экскреции β2-микроглобулина с мочой и снижение его уровня в крови. При межуточном нефрите сывороточная концентрация белка, определенная иммунохемилюминесцентным методом, не превышает 670 нг/мл, а его содержание в моче составляет более 300 мг/л.
- Пункционная биопсия почек. При остром процессе исследование биоптата позволяет обнаружить отек интерстиция, его инфильтрацию эозинофилами, плазмоцитами, мононуклеарные инфильтраты в перитубулярном пространстве, вакуолизацию канальцевого эпителия. О хроническом нефрите свидетельствует лимфоцитарная инфильтрация, атрофия канальцев и склероз стромы.
При хроническом интерстициальном воспалении наблюдается значительное снижение уровня эритроцитов и гемоглобина в общем анализе крови, при остром варианте нефрита возможна эозинофилия. Соответственно тяжести нарушений могут изменяться показатели электролитного баланса крови: увеличиваться или уменьшаться содержание калия, снижаться концентрации кальция, магния, натрия. При подозрении на возможную связь нефрита с системными заболеваниями дополнительно назначают анализы на выявление волчаночного антикоагулянта, антител к ds-ДНК, рибосомам, гистонам и другим нуклеарным компонентам. Часто определяется повышение уровней иммуноглобулинов — IgG, IgM, IgE.
Дифференциальная диагностика проводится между различными патологическими состояниями, которые осложняются интерстициальным воспалением. Заболевание также дифференцируют с острым, хроническим и быстропрогрессирующим гломерулонефритом, пиелонефритом, мочекаменной болезнью, опухолями почек. Кроме уролога и нефролога больным с подозрением на интерстициальный иммуновоспалительный процесс могут быть показаны консультации ревматолога, аллерголога-иммунолога, токсиколога, инфекциониста, фтизиатра, онколога, онкогематолога.
Лечение интерстициального нефрита
План ведения пациента определяется клинической формой и этиологическим фактором нефрологической патологии. Больных с симптоматикой острого межуточного нефрита экстренно госпитализируют в палату интенсивной терапии урологического или реанимационного отделения. При хроническом течении воспаления рекомендована плановая госпитализация в нефрологический стационар.
Основными терапевтическими задачами являются прекращение поступления и вывод из организма химического вещества, спровоцировавшего токсическое повреждение или гиперергическую иммуновоспалительную реакцию, десенсибилизация, детоксикация, стабилизация основного заболевания при вторичных формах нефрита, коррекция метаболических расстройств. С учетом стадии и течения болезни назначаются:
- Этиопатогенетическая терапия основного заболевания. Устранение причины, вызвавшей тубулоинтерстициальное воспаление, при отсутствии необратимых изменений канальцев и стромы позволяет быстрее нормализовать реабсорбционную и фильтрующую функции. При острых процессах, спровоцированных токсическими воздействиями, эффективны антидоты, энтеросорбенты, методы экстракорпоральной детоксикации. Грамотное лечение системных процессов направлено на предупреждение раннего развития ХПН.
- Иммуносупрессоры. При неэффективности детоксикационной терапии интерстициального медикаментозного нефрита, идиопатических формах заболевания, аутоиммунных болезнях часто применяют кортикостероиды в комбинации с антигистаминными средствами. Глюкокортикостероиды уменьшают отек межуточного вещества, ослабляют активность иммунного воспаления, противогистаминные препараты снижают выраженность гиперергического ответ. При дальнейшем нарастании симптоматики назначают цитостатики.
- Симптоматическое лечение. Поскольку острая почечная дисфункция зачастую сопровождается метаболическими расстройствами, пациентам с тубулоинтерстициальным нефритом показана интенсивная инфузионная терапия. Обычно под контролем диуреза вводят коллоидные, кристаллоидные растворы, препараты кальция. При аутоиммунных заболеваниях рекомендован прием антикоагулянтов, антиагрегантов.
- Для купирования возможной артериальной гипертензии используют блокаторы рецепторов ангиотензина.
При нарастании почечной недостаточности для предотвращения тяжелых уремических расстройств проводится заместительная терапия (перитонеальный диализ, гемодиализ, гемофильтрация, гемодиафильтрация). Больным с исходом хронического воспаления в выраженные склеротические изменения интерстициального вещества, атрофию канальцев и гломерул требуется трансплантация почки.
Прогноз и профилактика
При ранней диагностике и назначении адекватной этиотропной терапии полное выздоровление наступает более чем у 50% больных. Прогноз при интерстициальном нефрите благоприятный, если у пациента сохраняются нормальные показатели скорости гломерулярной фильтрации. Для предупреждения развития заболевания необходимо своевременное лечение инфекционных болезней почек, системных поражений соединительной ткани, ограничение приема нефротоксических препаратов (НПВС, антибиотиков из группы тетрациклина, петлевых диуретиков).
Меры индивидуальной профилактики нефрита включают употребление достаточного количества жидкости, отказ от самостоятельного приема лекарственных препаратов, прохождение регулярных медицинских осмотров, особенно при работе с производственными ядами.
1. Острый интерстициальный нефрит: значение морфологического диагноза/ Чуб О.И., Павленко Н.В., Мордовец Е.М.// Почки. – 2015 - №4 (14).
2. Клинические рекомендации по диагностике и лечению хронического тубулоинтерстициального нефрита. – 2017.
4. Острый тубулоинтерстициальный нефрит: проблемы диагностики/ Елисеева Л.И., Баранникова Е.И., Куринная В.П., Щербинина И.Г.// Нефрология и диализ – 2002 – Т.4, №2.
Нефронофтиз Фанкони
Нефронофтиз Фанкони – наследственное заболевание почек, характеризующееся поражением эпителия дистальных канальцев и петли нефрона, что приводит к нарушению выделительных функций и многочисленным вторичным патологиям. Симптомами этого состояния являются полиурия гипотонического характера, гипохромная анемия, поражения скелета, на терминальных этапах развивается азотемия. Диагностика нефронофтиза Фанкони включает в себя ультразвуковое исследование почек, анализы крови и мочи, молекулярно-генетические методы определения. Специфического лечения данного состояния не существует, используют паллиативную терапию, показана трансплантация почек.
Нефронофтиз Фанкони (нефронофтиз 1-го типа, ювенильный нефронофтиз) – генетическое заболевание, которое поражает почки (петли нефронов и дистальные канальцы) с развитием прогрессирующего нарушения выделительной функции данных органов. Впервые эта патология была описана в 1951-м году швейцарским педиатром Фанкони, после этого за все время наблюдений было выявлено несколько сотен случаев подобного состояния. Таким образом, нефронофтиз Фанкони является достаточно редким заболеванием, его встречаемость не определена. Кроме того, поражение почек, абсолютно идентичное данной патологии, встречается при ряде других генетических заболеваний – таких как синдром Жубера или синдром Сеньора-Локена 1-го типа. Однако при этих состояниях нарушения со стороны почек сочетаются с расстройствами функций других органов и систем (сетчаткой глаза, легких и пр.) – как удалось выяснить врачам-генетикам, схожесть патологий обусловлена тем, что их причиной выступают различные мутации одного и того же гена. Механизм наследования нефронофтиза Фанкони аутосомно-рецессивный, болезнь с одинаковой частотой встречается у мальчиков и у девочек. По разным оценкам, это заболевание является причиной 2-10% от всех случаев хронической почечной недостаточности в детском возрасте.
Причины нефронофтиза Фанкони
Нефронофтиз Фанкони обусловлен мутацией гена NPHP1, располагающегося на 2-й хромосоме. Продуктом экспрессии данного гена является особый белок под названием нефроцистин-1, функции которого в настоящее время остаются предметом бурных дискуссий. Согласно наиболее распространенной точке зрения, нефроцистин-1 входит в состав ресничек многих клеток – эпителия петель и канальцев почек и легких, содержится в сетчатой оболочке глаза и ряде других органов. В результате мутации NPHP1 нарушается структура данного протеина, что пагубно отражается на работе ресничек клетки, поэтому такие функции, как всасывание жидкости и растворов солей, а также активный транспорт различных ионов значительно нарушаются. Было установлено, что нефроцистин-1 способен связывать ряд других белков и ферментов, изменение его структуры, возможно, нарушает и эти процессы, приводя к развитию нефронофтиза Фанкони, синдромам Жубер или Сеньора-Локена.
По причине малоизученности функций белка нефроцистин-1 патогенез нефронофтиза Фанкони (как и других наследственных патологий, обусловленных дефектами гена NPHP1) также не совсем ясен. В частности, неизвестно, что больше нарушает процессы транспорта ионов и воды и образования мочи при этой патологии – изменение активности ресничек или неспособность нефроцистина-1 присоединять к себе другие белковые молекулы. Только начинает изучаться вопрос, какие мутации приводят к изолированному нефронофтизу Фанкони, а какие – к комплексным нарушениям. Достоверно известно, что первичными аномалиями при этом состоянии являются изменения в петлях нефрона и дистальных канальцах почек, все остальные симптомы (гипокальциемия, анемия, пороки развития скелета) считаются вторичными.
В результате дефекта гена NPHP1 в петлях нефрона и дистальных канальцах нарушаются процессы реабсорбции воды и неорганических солей, что приводит к увеличению объема образуемой мочи. По причине полиурии организм теряет огромное количество кальция и других ионов, что вызывает ответную реакцию паращитовидных желез (вторичный гиперпаратиреоз), гормон которых начинает вымывать кальций и фосфор из костной ткани. Элементы скелета становятся хрупкими, в них развиваются рахитоподобные изменения, которые очень часто составляют часть клинической картины нефронофтиза Фанкони. Патологические процессы затрагивают и юкстагломерулярный аппарат, в результате чего у больных развивается гипохромная анемия. Со временем нефронофтиз Фанкони приводит к прогрессирующей хронической почечной недостаточности, которая проявляется азотемией и другими сопутствующими нарушениями.
Симптомы нефронофтиза Фанкони
В большинстве случаев нефронофтиз Фанкони никак себя не проявляет в первые годы жизни ребенка, первые симптомы могут возникать в возрасте 2-3-х лет – наблюдаются полиурия и никтурия (увеличение доли ночного диуреза), в результате потери жидкости часто регистрируется жажда (полидипсия). Эти проявления выражены слабо и могут сохраняться на протяжении нескольких лет. Ухудшение при нефронофтизе Фанкони отмечается в возрасте 6-10 лет, когда вышеперечисленные симптомы становятся более выраженными и к ним присоединяются рахитоподобные изменения скелета, иногда – судороги, становится заметным отставание ребенка в физическом развитии. Часто обнаруживаются анемические проявления: бледность, тахикардия, головокружения и ряд других.
В дальнейшем при нефронофтизе Фанкони начинают нарастать признаки почечной недостаточности по тубулярному типу. По мере увеличения задержки азотистых продуктов обмена веществ в организме усиливается запах аммиака изо рта и от тела, развивается вторичный гиперпаратиреоз, обусловленный потерей значительного количества солей (в особенности, ионов кальция) с мочой. Все более выраженными становятся полиурия, полидипсия и другие нарушения работы почек. В конечном итоге нефронофтиз Фанкони приводит к тяжелой почечной недостаточности, требующей регулярного гемодиализа или трансплантации донорской почки. Иногда это состояние сочетается с врожденной глазодвигательной апраксией Когана, проявляющейся невозможностью движений глаз в горизонтальной плоскости у детей младше 2-3 лет.
Диагностика нефронофтиза Фанкони
Для определения нефронофтиза Фанкони применяют общие и биохимические анализы крови и мочи, ультразвуковое исследование почек, изучение наследственного анамнеза больного, молекулярно-генетические методы диагностики. Большое количество важной в диагностическом отношении информации дают такие методы, как определение клиренса эндогенного креатинина (проба Реберга), экскреторная урография, компьютерная томография почек с контрастом. При нефронофтизе Фанкони у больных выявляют увеличение суточного диуреза (что может быть установлено в ходе анализа мочи по Зимницкому), при этом плотность мочи снижена и в большинстве случаев равняется осмолярности плазмы крови (изостенурия). Из других показателей общего анализа мочи чаще всего регистрируют незначительную протеинурию (не более 1 г/сут), иногда возможна микрогематурия. На фоне этого у больных нефронофтизом Фанкони регистрируется сильная жажда, обусловленная потерей значительного объема жидкости.
Картина крови при нефронофтизе Фанкони включает в себя гипохромную анемию, гипокальциемию и значительное снижение концентрации других неорганических ионов, что является следствием так называемого сольтеряющего синдрома. Клиренс эндогенного креатинина неуклонно снижается по мере развития заболевания. На УЗИ почек у больных нефронофтизом Фанкони в начальной стадии наблюдается нарушение дифференцировки и разграничения коркового и мозгового вещества, в последнем возможно образование кист диаметром 2-8 миллиметров. В дальнейшем почки несколько уменьшаются в размерах, в них можно выявить признаки интерстициального фиброза.
При изучении наследственного анамнеза больного нередко обнаруживаются схожие клинические проявления у родственников, благодаря этому можно доказать аутосомно-рецессивный характер наследования. Генетическая диагностика нефронофтиза Фанкони производится методом прямого секвенирования гена NPHP1 для выявления мутаций (как правило, делеций или точечных). Кроме того, эта методика помогает дифференцировать заболевание от других комплексных синдромов, также обусловленных дефектами данного гена (например, синдрома Жубера). Методы современной генетики способны выявить скрытое носительство дефектного гена NPHP1, что может использоваться в рамках медико-генетического консультирования семьи перед зачатием ребенка.
Лечение и прогноз нефронофтиза Фанкони
Специфического лечения нефронофтиза Фанкони не существует, любые поддерживающие меры неспособны значительно увеличить выживаемость пациентов – как правило, эта патология приводит к тяжелой почечной недостаточности в возрасте до 20-ти лет. Единственным эффективным методом лечения заболевания является трансплантация почки, причем произведенная как можно раньше, до появления значительных необратимых вторичных нарушений. До момента проведения операции производят симптоматическое лечение нефронофтиза Фанкони, включающее солесодержащие растворы, обильное питье, препараты кальция. Для устранения анемии необходимо использовать экзогенные эритропоэтины – например, эритропоэтин-альфа. Если нефронофтиз Фанкони стал причиной почечной недостаточности, назначают гемодиализ, особую диету и сбалансированный водно-солевой режим.
Прогноз заболевания зависит от перспектив проведения трансплантации почки – при невозможности этой операции исход неблагоприятен, большинство больных умирает до 20-ти лет от нарастающей почечной недостаточности. Поддерживающая и симптоматическая терапия, а также использование гемодиализа, по мнению большинства специалистов, способны увеличить продолжительность жизни больного еще на несколько лет. Однако дать шанс на жизнь пациенту с нефронофтизом Фанкони способна только трансплантация почки. При принятии решения об операции оценивается множество факторов: общее состояние организма больного, наличие и выраженность вторичных нарушений, сопутствующие заболевания. После проведенной пересадки патологические изменения в донорской почке, аналогичные нефронофтизу Фанкони, не развиваются.
Сочетание нефронофтиза и аутосомной доминантной тубулоинтерстициальной болезни почек
Нефронофтиз и аутосомно-доминантное тубулоинтерстициальное заболевание почек (ADTKD) – наследственные заболевания, которые приводят к образованию кист исключительно в мозговом веществе почек или на границе коркового и мозгового вещества и впоследствии к терминальной стадии болезни почек.
Нефронофтиз и аутосомно-доминантное тубулоинтерстициальное заболевание почек (ADTKD) объединены в одну группу, поскольку имеют много общих характеристик. Их патогенез заключается в том, что кисты могут образовываться исключительно в мозговом веществе почки или на границе коркового вещества и мозгового, а также наблюдается триада: атрофия канальцев, дезинтеграция тубулярной базальной мембраны и интерстициальный фиброз. Кисты могут либо присутствовать, либо отсутствовать, и являются результатом трубчатой дилатации. Возможно, в их основе лежат одинаковые механизмы, однако они еще точно не установлены. Клинические признаки обоих заболеваний включают в себя следующее:
Вазопрессин (АДГ)-резистентное нарушение концентрационной способности почек, приводящее к полиурии и полидипсии
Достаточно существенная потеря натрия, требующая восполнения
Тенденция к умеренной полиурии и нормальный мочевой осадок
В итоге, терминальная стадия почечной недостаточности (ХПН)
Основные различия между нефронофтизом и медуллярной кистозной болезнью почек: механизм наследования и возраст дебюта хронической болезни почек Хроническая болезнь почек Хроническая болезнь почек (ХБП) представляет собой длительное прогрессивное снижение почечной функции. Симптомы развиваются медленно и на продвинутых стадиях включают в себя анорексию, тошноту. Прочитайте дополнительные сведенияНефронофтиз
Тип наследования – аутосомно-рецессивный. Нефронофтиз встречается в 15% случаев хронической болезни почек с почечной недостаточностью у детей и молодых взрослых < (моложе 20 лет). Существует 3 типа:
Детский, средний возраст начала – 1 год
Ювенильный, средний возраст начала – 13 лет
Подростковый, средний возраст начала – 19 лет
У пациентов с нефронофтизом описано одиннадцать генных мутаций. Наиболее часто встречаются мутации гена NPHP1, обнаруживаемые примерно у 30–60% пациентов. Около 10% пациентов с нефронофтизом имеют и другие клинические проявления, такие как пигментная дегенерация сетчатки, фиброз печени Фиброз печени Фиброз печени – это сверхвыраженный процесс заживления с образованием избыточного количества соединительной ткани, инкорпорированной в ткань печени. Происходит сверхпродукция экстрацеллюлярного. Прочитайте дополнительные сведения , снижение интеллекта и другие неврологические проблемы.
Терминальная стадия хронической почечной недостаточности (ТХПН) часто развивается в детстве и приводит к нарушениям роста и заболеваниям костей. Однако у многих пациентов эти проблемы развиваются медленно в течение многих лет и так хорошо компенсированы, что не кажутся патологическими, пока не появятся очевидные симптомы уремии Клинические проявления Хроническая болезнь почек (ХБП) представляет собой длительное прогрессивное снижение почечной функции. Симптомы развиваются медленно и на продвинутых стадиях включают в себя анорексию, тошноту. Прочитайте дополнительные сведения .
Диагностика нефронофтиза
Визуализирующие методы и/или генетическое исследование
Диагноз следует подозревать у детей со следующими симптомами (особенно при нормальном мочевом осадке):
Полидипсия и полиурия
Прогрессирующее ухудшение функции почек, особенно в отсутствие артериальной гипертензии
Сочетанная внепочечная патология
Анемия, степень которой не соответствует степени почечной недостаточности
Протеинурия, как правило, отсутствует. Диагноз подтверждается визуализирующими методами исследования, но кисты часто случаются только на поздней стадии заболевания. При УЗИ, КТ или МРТ можно выявить сглаженность контуров почек, их нормальные или уменьшенные размеры, отсутствие кортикомедуллярной дифференцировки и множество кист на границе коркового и мозгового вещества. Гидронефроз обычно отсутствует. Возможно генетическое исследование.
Лечение нефронофтиза
На ранней стадии заболевания лечение включает управление артериальной гипертензией Артериальная гипертензия Артериальная гипертензия – это стойкое повышение систолического артериального давления в покое (≥ 130 мм рт.ст.) и/или диастолического артериального давления (≥ 80 мм рт.ст.). Повышение АД без. Прочитайте дополнительные сведения и им требуется проведение диализа Гемодиализ При гемодиализе кровь пациента подается в диализатор, содержащий 2 жидкостных компартмента, формируемых последовательно или параллельно соединенными полыми капиллярными волокнами и листками. Прочитайте дополнительные сведения или трансплантация Трансплантация почки Трансплантация почек является наиболее распространенным видом трансплантации цельных органов. (См. также Обзор трансплантации (Overview of Transplantation)). Основное показание для трансплантации. Прочитайте дополнительные сведения почки.
Аутосомно-доминантная тубулоинтерстициальная болезнь почек (ADTKD)
Аутосомно-доминантная тубулоинтерстициальная болезнь почек (медуллярная кистозная болезнь почек) представляет собой группу редко встречающихся генетических нарушений. Консенсусный отчет ( 1 Справочные материалы по лечению Нефронофтиз и аутосомно-доминантное тубулоинтерстициальное заболевание почек (ADTKD) – наследственные заболевания, которые приводят к образованию кист исключительно в мозговом веществе почек. Прочитайте дополнительные сведения ) от "Болезни почек: Улучшения мировых результатов лечения (KDIGO)" предложил классификацию этих заболеваний на основе причинного гена, из которых 4 в настоящее время известны (см. таблицу Аутосомно-доминантная тубулоинтерстициальная болезнь почек: классификация на основе генетического анализ а).
Нефронофтиз - типы, диагностика, лечение
Выделяют три формы нефронофтиза — все с аутосомно-рецессивным типом наследования. Нефронофтиз типа II начинается в грудном возрасте, нефронофтиз типа III впервые проявляется у подростков, а начало нефронофтиза типа I (ювенильного нефронофтиза) приходится на препубертатный период. Иногда ювенильный нефронофтиз протекает с внепочечными проявлениями.
Нефронофтиз типа I (ювенильный нефронофтиз)
Для детей с ювенильным нефронофтизом характерны полиурия и полидипсия, анемия и задержка роста. Поскольку заболевание не проявляется типичными признаками гломерулопатии (гематурией, протеинурией, артериальной гипертонией, отеками), почечную недостаточность нередко обнаруживают случайно при обследовании по поводу анемии, полиурии или сонливости. Диагноз нефронофтиза в таких случаях может оказаться полной неожиданностью. Средний возраст развития терминальной почечной недостаточности — 13 лет.
Внепочечные проявления нефронофтиза. Ювенильный нефронофтиз может сопровождаться врожденной глазодвигательной апраксией Когана, при которой у гомозиготных по делециям гена NPHP1 детей младше 2 лет отсутствуют произвольные движения глаз по горизонтали. Вместе с пигментной дегенерацией сетчатки нефронофтиз входит в состав синдрома Сениора—Локен, а вместе с агенезией червя мозжечка и колобомой диска зрительного нерва - в состав синдрома Жубер типа II. Описан также нефронофтиз в сочетании с фиброзом печени и с конусовидными эпифизами фаланг.
Наконец, ювенильный нефронофтиз может быть одним из проявлений синдромов Жена (асфиктическая дисплазия грудной клетки) и Эллисаван Кревельда (хондроэктодермальная дисплазия), синдрома RHYNS (Retinitis pigmentosa, HYpopituitarism, Nephronophthisis, mild Skeletal dysplasia — пигментная дегенерация сетчатки, гипопитуитаризм, нефронофтиз, легкие аномалии развития скелета), а также синдромов Лоренса—Муна и Барде—Бидля.
Диагностика нефронофтиза. Важный метод диагностики нефронофтиза — УЗИ, при котором выявляются сглаженность границы между корковым и мозговым веществом, повышенная эхогенность почечной паренхимы, а у детей старше 9 лет — кисты, расположенные на границе коркового и мозгового вещества. Сцинтиграфия обнаруживает снижение концентрирующей способности почек.
Нефронофтиз типа II
Этот тип нефронофтиза называют инфантильным, так как первые его проявления возникают еще во внутриутробном периоде, сразу после рождения или в первый год жизни ребенка. Недавно при исследовании родословной большой семьи бедуинов было установлено, что ген нефронофтиза типа II (NPHP2) находится на 9-й хромосоме (сегмент q22-q31).
Поскольку морфологическая картина и течение этого заболевания довольно сильно отличаются от двух других форм нефронофтиза, возможно, что в группу нефронофтиза оно попало не совсем обоснованно.
Нефронофтиз типа III
Ген нефронофтиза типа III (NPHP3) идентифицирован при исследовании еще одной большой родословной — на этот раз семьи из Венесуэлы. Этот тип нефронофтиза называют подростковым. Терминальная почечная недостаточность наступает на 6 лет позже, чем у больных ювенильным нефронофтизом, — в среднем в 19 лет. В остальном же ни морфологической картиной, ни клиническими проявлениями нефронофтиз типа III от ювенильного нефронофтиза не отличается.
Генодиагностика нефронофтиза
Идентификация гена NPHP1, ответственного за ювенильный нефронофтиз, позволила использовать для диагностики прямой анализ ДНК. Продукт этого гена нефрокистин содержит SH3-домен, что, по-видимому, указывает на участие нефрокистина в межбелковых взаимодействиях, в том числе во внутриклеточной передаче сигнала в фокальных контактах — участках, в которых клетка соприкасается с внеклеточным матриксом.
Нарушением фокальных контактов могут объясняться характерные разрывы базальной мембраны почечных канальцев, наблюдаемые при нефронофтизе. У 85% детей делеции обнаруживаются на обоих аллелях гена NPHP1. На выявлении этих делеций, а также точечных мутаций гена NPHP1 основывается генодиагностика нефронофтиза. Гомозиготные делеции гена NPHP1 описаны также при врожденной глазодвигательной апраксии Когана и при поздней форме пигментной дегенерации сетчатки.
При глазодвигательной апраксии Когана детям грудного и младшего возраста не удаются горизонтальные движения глаз и, чтобы посмотреть в сторону, они совершают резкие толчкообразные движения головой.
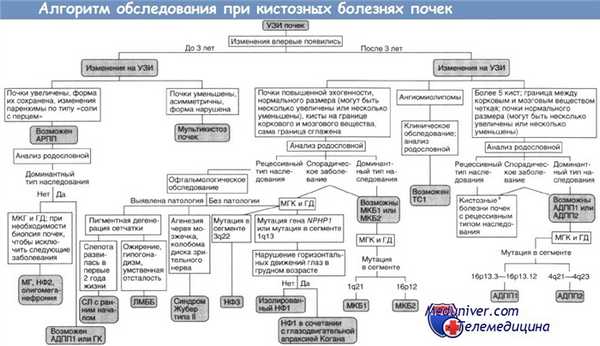
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Читайте также:
- Лечение заворота сигмовидной кишки. Консервативное и оперативное лечение заворота сигмовидной кишки.
- Влияние толуола на плод и беременность
- Кровоснабжение легких. Лимфоотток от легкого. Иннервация легких.
- Как выполнить катетеризацию внутренней яремной вены
- Операция при травматическом шоке. Диагностика полостных кровотечений