Врожденный фибросаркомоподобный фиброматоз. Врожденный генерализованный фиброматоз.
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Инфантильная фибросаркома (ИФ) – редкая саркома мягких тканей, возникающая с частотой 1 % всех случаев злокачественных новообразований у детей на первом году жизни. ИФ чаще всего поражает мягкие ткани верхних и нижних конечностей. К редким локализациям относят туловище, область головы и шеи и желудочно-кишечный тракт. В статье описан редкий клинический случай врожденной ИФ мягких тканей грудной стенки у новорожденного, а также литературный обзор.
Ключевые слова
Об авторах
НИИ детской онкологии и гематологии ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России
Россия
Гарик Барисович Сагоян, врач-детский онколог
115478, Москва, Каширское шоссе, 23
к.м.н., старший научный сотрудник 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
д.м.н., ведущий научный сотрудник 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
врач-детский онколог 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
к.м.н., врач-детский хирург 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
врач-детский онколог 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
115478, Москва, Каширское шоссе, 23
врач-детский онколог 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
врач-детский хирург 2-го хирургического отделения
115478, Москва, Каширское шоссе, 23
д.м.н., заведующая отделением анестезиологии-реанимации
115478, Москва, Каширское шоссе, 23
к.м.н., заведующая рентгенодиагностическим отделением отдела лучевых методов диагностики и лечения опухолей
115478, Москва, Каширское шоссе, 23
д.м.н., заведующий 2-м хирургическим отделением
115478, Москва, Каширское шоссе, 23
д.м.н., профессор, заместитель директора по научной и лечебной работе – директор НИИ детской онкологии и гематологии аппарата управления НМИЦ онкологии им. Н.Н. Блохина, исполнительный директор РОО НОДГО
115478, Москва, Каширское шоссе, 23
Список литературы
1. Kaatsch P., Spix C. German Childhood Cancer Registry – Annual Report 2015 (1980–2014). Institute for Medical Biostatistics, Epidemiology and Informatics (IMBEI), at the University Medical Center of the Johannes Gutenberg University Mainz, 2015.
2. Cecchetto G., Carli M., Alaggio R., Dall’Igna P., Bisogno G., Scarzello G., Zanetti I., Durante G., Inserra A., Siracusa F., Guglielmi M.; Italian Cooperative Group. Fibrosarcoma in pediatric patients: results of the Italian cooperative group studies (1979–1995). J Surg Oncol 2001;78:225–31. doi: 10.1002/jso.1157.
3. Coffin C.M., Dehner L.P. Soft tissue tumors in first year of life: a report of 190 cases. Pediatr Pathol 1990;10:509–26. doi: 10.3109/15513819009067140.
4. Zeytun H., Okur M.H., Basuguy E., Arslan S., Aydogdu B., Turkcu G., Arslan M.S. Congenital-infantile fibrosarcoma of the ileocecal region: the first case presentation. Pediatr Surg Int 2016;32(1):97–9. doi: 10.1007/s00383-015-3802-0.
5. Gurney J.G., Young J.L., Roffers S.D., Smith M.A., Bunin G.R. Soft tissue sarcomas. In: Gloecker Ries I.A., Smith M.A., Gurney J.G., Linet M., Tamra T., Young J.L., Bunin G.R., eds. SEER pediatric monograph: cancer incidence and survival among children and adolescents, United States SEER program 1975–1995. National Cancer Institute, Bethesda, 1999. Pp. 111–124.
6. Parida L., Fernandez-Pineda I., Uffman J.K., Davidoff A.M., Krasin M.J., Pappo A., Rao B.N. Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 2013;29(7):703–8. doi: 10.1007/s00383-013-3326-4.
8. Ferguson W.S. Advances in the adjuvant treatment of infantile fibrosarcoma. Exp Rev Anticancer Ther 2003;3:185–91. doi: 10.1586/14737140.3.2.185.
9. Coffin C.M., Dehner L.P. Fibroblastic-myofibroblastic tumors in children and adolescents clinicopathologic study of 108 examples in 222 patients. Pediatr Pathol 1991;11(4):569–88. doi: 10.3109/15513819109064791.
11. Pandey A., Kureel S.N., Bappavad R.P. Chest Wall Infantile Fibrosarcomas – A Rare Presentation. Indian J Surg Oncol 2016;7(1):127–9. doi: 10.1007/s13193-016-0487-3.
12. van Grotel M., Blanco E., Sebire N.J., Slater O., Chowdhury T., Anderson J. Distant metastatic spread of molecularly proven infantile fibrosarcoma of the chest in a 2-month-old girl: case report and review of literature. J Pediatr Hematol Oncol 2014;36(3):231–3. doi: 10.1097/MPH.0000000000000055.
13. Shamberger R.C., Grier H.E., Weinstein H.J., Perez-Atayde A.R., Tarbell N.J. Chest wall tumors in infancy and childhood. Cancer 1989;63(4):774–85. doi: 10.1002/1097-0142(19890215)63:43.0.co;2-2.
14. Sultan I., Casanova M., Al-Jumaily U., Meazza C., Rodriguez-Galindo C., Ferrari A. Soft tissue sarcomas in the first year of life. Eur J Cancer 2010;46(13):2449–56. doi: 10.1016/j.ejca.2010.05.002.
15. Coffin C.M., Fletcher J.A. Infantile fibrosarkoma. In: Fletcher С.D., Unni K.K., Mertens F., eds. WHO classification of tumours. Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2002. Pp. 98–100.
19. Loh M.L., Ahn P., Perez-Atayde A.R., Gebhardt M.C., Shamberger R.C., Grier H.E. Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children’s Hospital, Boston. J Pediatr Hematol Oncol 2002;24(9):722–6. doi: 10.1097/00043426-200212000-00008.
20. Orbach D., Brennan B., De Paoli A., Gallego S., Mudry P., Francotte N., van Noesel M., Kelsey A., Alaggio R., Ranchère D., De Salvo G.L., Casanova M., Bergeron C., Merks J.H., Jenney M., Stevens M.C., Bisogno G., Ferrari A. Conservative strategy in infantile fibrosarcoma is possible: the European Paediatric Soft Tissue Sarcoma Study Group experience. Eur J Cancer 2016;57:1–9. doi: 10.1016/j.ejca.2015.12.028.
22. Minard-Colin V., Orbach D., Martelli H., Bodemer C., Oberlin O. Soft tissuetumors in neonates. Arch Pediatr 2009;16(7):1039–48. doi: 10.1016/j.arcped.2009.03.005.
24. Garber K. In a maior shift, cancer drugs go “tissue-agnostic”. Science 2017;356(6343):1111–2. doi: 10.1126/science.356.6343.1111.
25. Laetsch T.W., DuBois S.G., Mascarenhas L., Turpin B., Federman N., Albert C.M., Nagasubramanian R., Davis J.L., Rudzinski E., Feraco A.M., Tuch B.B., Ebata K.T., Reynolds M., Smith S., Cruickshank S., Cox M.C., Pappo A.S., Hawkins D.S. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018;19(5):705–14. doi: 10.1016/S1470-2045(18)30119-0.
Фиброматоз дёсен (гипертрофия десны): причины, диагностика, как предотвратить
Фиброматоз десен – неконтролируемое разрастание фиброзной ткани в области десен и пародонта. Может быть наследственным или приобретенном, отличается прогрессирующим характером и требует хирургической коррекции. В отсутствие лечения разрастание тканей неминуемо приведет к развитию сопутствующих патологий с образованием зубодесневых карманов, атрофией костной ткани, утратой здоровых зубов и нарушению жевательной функции. У детей наблюдается нарушение роста зубов и выраженное искажение прикуса.
Почему возникает фиброматоз десен
Причины развития заболевания:
генетическая патология – одиночные мутации гена, некоторые формы синдрома Дауна;
длительный прием некоторых лекарственных препаратов – противоэпилептических средств, иммуносупрессоров;
гормональные нарушения –- заболевания эндокринной системы, длительный прием гормональных контрацептивов, анаболических стероидов и т.п.
Спровоцировать разрастание тканей может и хирургическое вмешательство.
Кроме того, сходные с фиброматозом симптомы проявляются при некоторых видах хронического инфекционного стоматита.
Генетически обусловленная форма гипертрофии десны диагностируется после протезирования зубов и может длиться всю жизнь, требуя периодического лечения. Лекарственная и гормональная формы могут появляться независимо от пола и возраста пациента и полностью проходят после устранения причины: например, отмены или замены препарата, лечения эндокринного заболевания.
Как проявляется болезнь
Разрастание десен затрагивает внешнюю (щечную) и внутреннюю (языковую) область десен с охватом одной или обеих челюстей. Поражаются общая поверхность десны, прилегающий к зубу десневой край, а также межзубные сосочки.
Основные симптомы патологии:
частичное или полное зарастание коронок в области фиброматоза;
гладкий округлый край разрастаний;
повышенная плотность гипертрофированных тканей;
ровный розовый оттенок десны и отсутствие кровоточивости.
Интенсивность проявления симптомов определяет стадию заболевания.
При этом гипертрофия десен может быть генерализованной или очаговой. Разница заключается в том, что если при очаговом формируется только единичный очаг поражения, то при генерализованной форме несколько патологических участков сливаются между собой. В тяжелых случаях проявления болезни фиброматоз может сочетаться с изменением черт лица, характерным для акромегалии.
На заметку! При наследственной патологии повернуть вспять гипертрофию тканей невозможно, однако на участках десны с удаленными зубами разрастание обычно приостанавливается.
Диагностика фиброматоза десен
Обследование включает осмотр ротовой полости в кабинете стоматолога. Слизистые оболочки проверяют на болезненность, цветовой оттенок, плотность тканей и их уровень относительно коронки зуба.
Фиброматоз относят к опухолевым заболеваниям, поэтому в ходе обследования важно дифференцировать его с другими видами новообразований. Для этого назначают дополнительную диагностику:
Биопсия и гистологический анализ тканей позволяют отделить болезнь от сходных проявлений полиомиелита, пародонтита, рака.
Рентген оценивает состояние альвеолярных отростков костной ткани и структуру челюстей в целом.
Фиброматоз десен: лечение
Для успешной терапии лекарственной и гормональной форм заболевания достаточно устранения основной причины (замена лекарства, ликвидация дисбаланса гормонов). Основной способ лечения наследственной гипертрофии десен – регулярное хирургическое воздействие скальпелем или лазером. При этом каждый курс оперативного вмешательства проводится поэтапно и занимает несколько недель.
Последовательность хирургических манипуляций:
введение анестезии – инфильтрационной или проводниковой;
иссечение фиброзных разрастаний;
удаление всех шатающихся зубов (при необходимости);
антисептическая обработка раны и наложение специальной мембраны.
Процесс заживления занимает 1–2 недели, после чего процедуру повторяют на другой челюсти. Периодичность проведения процедур индивидуальна и зависит от скорости прогрессирования фиброматоза.
Профилактика
Профилактику фиброматоза нужно проводить в течение всего периода роста и формирования прикуса у детей. Основное внимание следует уделять гигиене ротовой полости, в том числе удалению зубных отложений. Дополнительно следует избегать травматизации слизистых оболочек грубой пищей или жесткими зубными щетками. Регулярное посещение кабинета стоматолога позволит своевременно выявить первые признаки болезни и предупредить осложнения заболевания.
Врожденный фибросаркомоподобный фиброматоз. Врожденный генерализованный фиброматоз.
Врожденный фибросаркомоподобный фиброматоз. Врожденный генерализованный фиброматоз.
Врожденный фибросаркомоподобный фиброматоз (врожденный фиброматоз, врожденная фибросаркома, фибросаркома младенцев, медуллярный фиброматоз, врожденный агрессивный фиброматоз, агрессивный инфантильный фиброматоз). Р. W. Allen вслед за A. Balsaver и соавт. (1967) обратил внимание на поразительное сходство этих поражений, имеющих врожденный характер или возникающих в первые 3 мес жизни, с фибросаркомой взрослых при отсутствии метастазов. Новообразования локализуются на голове, в подмышечной области, руке, спине, бедре, голене и стопе у лиц обоего пола. Узлы достигают 8,5 см в диаметре, часто возвышаются над кожей, изъязвляются. обычно плохо отграничены, инфильтрируют кожу, подкожную клетчатку и мышцы. Микроскопически узлы представлены веретенообразными фибробластоподобными клетками с явлениями гиперхроматоза и полиморфизма ядер. Образования обычно имеют большое количество клеток, с частыми фигурами митоза и четким рисунком «елочки». Иногда наблюдают телеангиэктазии с эндотелиальной пролиферацией, фокусы некроза, окруженные палисадообразно располагающимися клетками, участки выраженного коллагенообразоваиия, инфильтрация лимфоидными элементами. Рецидивы после хирургического иссечения возникают приблизительно в половине наблюдений.
Нам представляется, что ценность временного критерия (возникновение опухоли в первые 3 мес жизни) при отсутствии других более важных (клинических и морфологических) критериев для выделения этой формы фиброматозов весьма спорна. Поэтому целесообразно диагностировать все подобные поражения как врожденную фибросаркому и ограничиваться диагностической дилеммой врожденная фибросаркома — врожденный фибросаркомоподобный фиброматоз.
Окончательное решение вопроса о характере процесса требует новых исследований с новыми методическими подходами.
Врожденный генерализованный фиброматоз (врожденный мультицентричный фиброматоз, врожденный множественный фиброматоз, врожденная фибросаркома, множественная сосудистая лейомиома новорожденных, множественные врожденные мезенхимальные опухоли) впервые был описан А. Р. Stout (1954) как опухоль с поражением подкожной клетчатки, глубоких мягких тканей, костей и внутренних органов Встречается редко
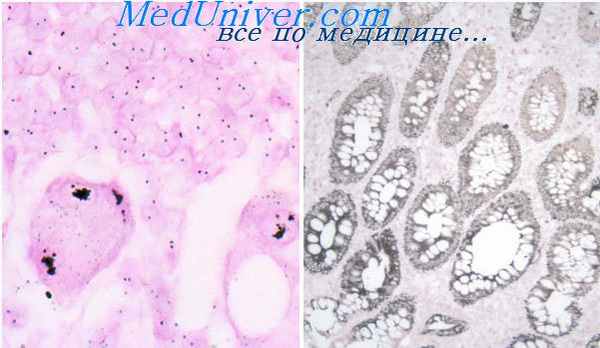
Уже при рождении визуально и пальпаторно определяют поверхностно-расположенные узлы; глубокорасположенные выявляют рентгенологически или обнаруживают при аутопсии Число узлов может доходить до 51, они быстро увеличиваются в объеме, появляются новые, кожа над крупными узлами может изъязвляться. Отмечается некоторая наклонность к поражению туловища, бедра, плечевого пояса. Глубокие очаги располагаются в сердце, легких, плевре, кишечнике, брюшине, языке, гортани, печени, поджелудочной железе, костях, где сочетаются с остеолизом В 1981 г Е В Chung и F M Enzinger сообщили о единственном пока наблюдении врожденного генерализованного фиброматоза с множественным поражением ЦНС С поражением тех или иных органов связана и симптоматика, и причина смерти, которой закончились почти все описанные наблюдения. В некоторых случаях к смерти приводят интеркуррентные инфекции.
Макроскопически большинство узлов хорошо отграничено, размер узлов редко превышает 2,5 см, они плотные и эластичные, округлые, на разрезе серого цвета, часто с очагами некроза, дегенерации, кровоизлияний
Микроскопически эти поражения обычно с умеренным количеством клеток, это «пухлые» веретенообразные клетки со скудной цитоплазмой, имеющие сходство с гладкомышечиыми элементами, богатые сосудами Часто встречаются центральные некрозы с гиалинозом и кальцинацией. В таких участках тени клеток вместе с тонкостенными сосудами часто образуют «гемангиоперицитомоподобный» рисунок. Коллагенообразованне, как правило, незначительно
Подобные поражения следует дифференцировать с врожденным неврофиброматозом, врожденной злокачественной шваииомой на фоне нейрофиброматоза, множественной врожденной гемангиоперицитомой, множественными врожденными лейомиомами, врожденной лейомиосаркомой или фибросаркомой с множественными метастазами. Большинство детей умирают вскоре после рождения. Описаны случаи регрессии процесса, если ребенку удавалось пережить "критические" первые 5 нед жизни.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Врожденный фибросаркомоподобный фиброматоз. Врожденный генерализованный фиброматоз.
Ювенильная назофарингеальная фиброма (снн. ювенильная ангиофиброма носоглотки) — редкая форма, поражающая, главным образом, лиц мужского пола в возрасте 10— 25 лет. Плотноватый серый или красноватый узел диаметром 2—5 см растет в верхней части заднебоковой стенки полости носа в зоне сочленений клиновидного отростка небной кости, крыловидного отростка клиновидной кости и горизонтального крыла сошника (в перегородке носа). Опухоль склонна к рецидиву it иногда к спонтанной регрессии. Под микроскопом в плотной или рыхлой, иногда отечной строме располагаются в довольно большом количестве мелкие тонкостенные и крупные сосуды.
Ювенильную назофарингеальную фиброму нужно отличать от полипа носовой перегородки и гемангиомы.
Инфантильныий миофиброматоз
Инфантильный миофиброматоз (син: врожденный генерализованный, множественный мультицентрический фиброматоз; инфантильная гемангиоперицитома; врожденная фибросаркома; агрессивный инфантильный фиброматоз: множественные врожденные мезенхнмапьные опухоли: множественная сосудистая лейомиома новорожденных) — одиночный узел (2/3 наблюдений) или множественное поражение (1/3) глубоких мягких тканей новорожденных, детей грудного возраста, реже детей старше 2 лет, крайне редко юношей и взрослых лиц. Локализация: подкожная клетчатка, мышцы и, кроме того, при инфантильном генерализованном варианте — кости, легкие, сердце, желудочно-кишечный тракт и другие внутренние органы.
При инфантильном мультицентрическом варианте с поражением мягких тканей и костей встречается спонтанное самоизлечение. Макроскопически узел обычно имеет Рубцовую консистенцию, диаметр до 5 см, четкие границы, иногда капсулу и петрификаты. Под микроскопом периферические зоны узла сформированы из хаотично переплетающихся пучков миофиб-робластов, центральные зоны — из пучков более мелких, незрелых и делящихся фибробластов. Часто развиты мелкие ветвящиеся сосуды синусоидного типа. Незрелые формы поражения характерны для младенцев, а формы, близкие к десмоиду, — для взрослых лиц. Инфантильный миофиброматоз следует отличать от гемангиоперицитомы, десмоидного фиброматоза, нейрофиброматоза, врожденных лейомиом, лейомиосарком, а также фибросарком и гамартом.
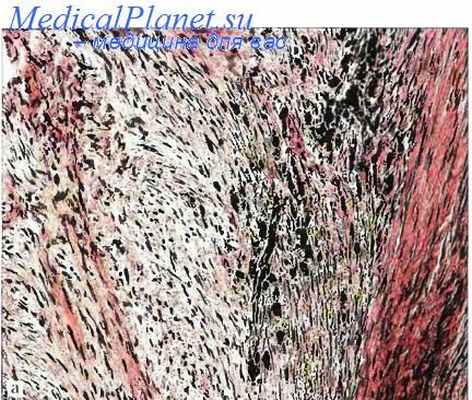
Инфантильный миофиброматоз
Воспалительная миофибробластическая опухоль
Воспалительная миофибробластическая опухоль (син.: воспалительная псевдоопухоль, воспалительная фибросаркома, миофибробластома, смеша нноклеточная псевдоопухоль, фибром и ксо-идная лимфоплазмогистиоцитома) — групповое обозначение довольно редких опухолей, поражающих лиц обоего пола и разного возраста, зачастую детей. Различают легочную и внелегочную формы. В последнем случае поражаются мягкие ткани конечностей, селезенка, забрюшинные и медиастинальные мягкие ткани, кожа, урогенитальная система, желудочно-кишечный тракт и др. Макроскопически представляют собой четко отграниченный, плотноватый, беловато-серый или пестрый узел диаметром от 2 до 20 см. Бывают множественные узлы. Опухоль склонна к рецидивам. Описаны случаи малигнизации (трансформация в саркому). Под микроскопом определяется воспалительный компонент, который сопровождает три разных варианта пролиферации миофибробластов. Первый вариант напоминает обычную грануляционную ткань или нодозный фасциит.
Вытянутые миофибробласты с везикулярными ядрами лежат в рыхлой или миксоидной строме вперемешку с нейтрофилами, лимфоцитами, эо-зинофилами и более редкими плазматическими клетками. Второй вариант отличается большей, но очаговой «клеточностью», представленной веретеновидными фибробластами и миофибробластами в компактной строме. «Клеточные» очаги окружены миксоидной или гиалинизированной стромой. В них встречаются фигуры митоза, большое количество плазматических клеток и лимфоцитов, местами формирующих фолликулы. Третий вариант проявляется в сильном развитии гиалинизированной и малоклеточной стромы с небольшим количеством плазматических клеток и лимфоцитов. Изредка встречаются гистиоцитоподобные или ганглиозно-подобные клетки с крупными везикулярными ядрами и развитыми ядрышками. В 24—93 % случаев опухоль позитивна в отношении CD68, в ней отмечаются хромосомные аберрации 2р23. Воспалительную миофибробластическую опухоль следует отличать от нодозного фасциита, серозитов, выбухающей дерматофибромы, злокачественной фиброзной гистиоцитомы, различных сарком, лимфом, болезни Розаи—Дорфмана и плазмоцитомы.
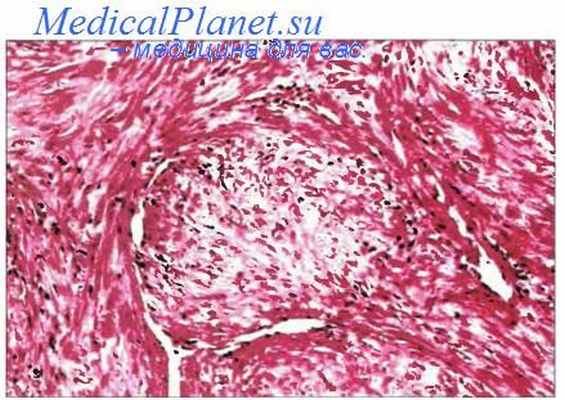
Инфантильный миофиброматоз
Воспалительная миксоглалиновая опухоль конечностей
Воспалительная миксоглалиновая опухоль конечностей (син. акральная миксовослалительная фибробластная саркома) — весьма редкое новообразование со слабо выраженным злокачественным потенциалом. Пол и возраст больных значения не имеют. Локализация: в большинстве случаев верхние, иногда нижние конечности. Медленно растущий, дольчатый фиброзный узел плотной или желатиновой консистенции, диаметром 1—8 см, располагается обычно в подкожной клетчатке. Отмечена склонность к рецидивам и к метастазированию в регионарные лимфатические узлы. Под микроскопом видны узелки миксоидной ткани, окруженные стромой, богатой атипичными веретеновидными и эпителиоидными клетками. Отмечаются также различное количество элементов воспалительного инфильтрата и повсеместное наличие крупных атипичных клеток с причудливыми ядрами и развитыми ядрышками. Эти клетки напоминают либо ганглиозные клетки, либо, в случае расщепленного ядра, клетки Рид—Штернберга. Они отличаются экспрессией виментина, CD68 и CD34. Изредка обнаруживаются фигуры митоза и отложения гемосидерина. Опухоль следует дифференцировать от теносиновита, воспалительной миофибробластической опухоли, юкстаартикулярной миксомы, миксофибросаркомы.
ОПУХОЛИ МЯГКИХ ТКАНЕЙ
мед.
Заболеваемость. Саркомы мягких тканей составляют 1 % всех злокачественных новообразований у взрослых. Опухоли в равной степени поражают мужчин и женщин, чаще в возрасте 20-50 лет. Возможно возникновение в детском возрасте (10-11% сарком). Гистогенез. Источник опухолей — самые разнородные по строению и происхождению ткани. В основном, это производные мезенхимы: фиброзная соединительная, жировая, синовиальная и сосудистая ткани, а также ткани, связанные с мезодермой (поперечно-полосатые мышцы) и нейроэктодермой (оболочки нервов). Гистогенетическая классификация
• Мезенхима:
• Злокачественная мезенхимома
• Миксома
• Фиброзная ткань:
• Десмоид (инвазивная форма)
• Фибросаркома
• Жировая ткань — липосаркома
• Сосудистая ткань:
• Злокачественная гемангиоэндотелиома
• Злокачественная гемангиоперицитома
• Злокачественная лимфангиосаркома
• Мышечная ткань:
• Поперечно-полосатые мышцы — рабдомиосаркома
• Гладкие мышцы — лейомиосаркома
• Синовиальная ткань — синовиальная саркома
• Оболочки нервов:
• Нейроэктодермальные — злокачественная невринома (шваннома)
• Соединительнотканные — периневральная фибросаркома
• Неклассифицируемые бластомы.
Факторы риска
• Радиоактивное облучение
• Действие химических веществ (например, асбеста или древесных консервантов)
• Генетические нарушения. Например, у 10% пациентов с болезнью фон Реклингхаузена развивается нейрофибросаркома
• Предшествующие заболевания кости. У 0,2% пациентов с болезнью Пёджета (деформирующий остоз) развиваются остеосаркомы.
Клиническая картина
• Мягкотканные саркомы. Разрастание ткани, наличие опухоли, боль в туловище или конечностях, но само образование безболезненно, на разрезе напоминает рыбье мясо
• Забрюшинные опухоли. Больные обычно отмечают снижение массы тела и предъявляют жалобы на боли неопределённой локализации. Опухоли могут достигать значительных размеров без клинических проявлений
• Кровотечение — самое частое проявление сарком ЖКТ и женских половых органов.
Диагностика
• Биопсия
• Быстро растущая опухоль (или разрастание ткани, превышающее в диаметре 5 см) скорее всего злокачественна, особенно если она плотная, спаяна с окружающими тканями и глубоко расположена
• При подобных поражениях необходима эксцизионная биопсия; пункционная биопсия чаще всего неинформативна
• При выборе места для биопсии следует учитывать возможное проведение в последующем реконструктивной (пластической) операции
• Радиологическое обследование: рентгенография, сцинтиграфия костей, МРТ, КГ
• Для мягкотканных сарком предпочтительнее МРТ-диагностика, обеспечивающая более точное определение границы между опухолями и мягкими тканями
• КГ и сцинтиграфия костей предпочтительнее для обнаружения костных поражений
• При признаках нарушения функций печени при саркомах внутренних органов или конечностей проводят КГ и УЗИ (для выявления метастазов)
• При подозрении на прорастание сосудов показана контрастная ангиография.
Лечение
Читайте также: