
Болезнь ниманна пика презентация
Презентация была опубликована 6 лет назад пользователемНадежда Шумилова
Презентация на тему: " Болезнь Нимана-Пика Выполнила: студентка 5 курса 5 группы, 4 мед. Ф-та Новосельцева О.М." — Транскрипт:
1 Болезнь Нимана-Пика Выполнила: студентка 5 курса 5 группы, 4 мед. Ф-та Новосельцева О.М.
2 Определение Болезнь Ниманна - Пика - наследственное заболевание, при котором дефицит специфического фермента приводит к накоплению сфингомиелина (продукта расщепления жиров). Различают более пяти форм болезни Ниманна - Пика в зависимости от того, насколько выражен дефицит фермента. Болезнь Ниманна - Пика - наследственное заболевание, при котором дефицит специфического фермента приводит к накоплению сфингомиелина (продукта расщепления жиров). Различают более пяти форм болезни Ниманна - Пика в зависимости от того, насколько выражен дефицит фермента.
4 Классификация В 1961 году, была предложена следующая классификация заболевания: В 1961 году, была предложена следующая классификация заболевания: - болезнь Ниманна-Пика, тип А: классическая инфантильная; - болезнь Ниманна-Пика, тип А: классическая инфантильная; - болезнь Ниманна-Пика, тип В: висцеральная; - болезнь Ниманна-Пика, тип В: висцеральная; - болезнь Ниманна-Пика, тип С: неострая подростковая - болезнь Ниманна-Пика, тип С: неострая подростковая - болезнь Ниманна-Пика, тип D: ново-шотландская; - болезнь Ниманна-Пика, тип D: ново-шотландская;
5 Однако, на сегодня, когда понятна генетическая природа заболевания, расстройство классифицируется следующим образом: Однако, на сегодня, когда понятна генетическая природа заболевания, расстройство классифицируется следующим образом: - болезнь Ниманна-Пика, связанная с геном SMPD1, которая включает в себя типы А и В; - болезнь Ниманна-Пика, связанная с геном SMPD1, которая включает в себя типы А и В; - болезнь Ниманна-Пика, типа C, который включает в себя типы C1 и C2. (Тип D возникает в результате мутации того же гена, что и тип C1). - болезнь Ниманна-Пика, типа C, который включает в себя типы C1 и C2. (Тип D возникает в результате мутации того же гена, что и тип C1).
6 Этиология Два типа болезни Ниманна-Пика, А и В возникают вследствие мутаций гена SMPD1, в то время как болезнь Ниманна-Пика типа С, вызывают мутации, происходящие в генах NPC1 и NPC2. Ранее, был еще один тип болезни - тип D, который использовался для того, чтобы отделить тех больных, которые происходили из Новой Шотландии. Ведь у этих лиц заболевания возникает из-за мутации гена NPC1. Однако, на сегодня такое разделение считают нецелесообразным и тип С включает в себя обе группы мутаций. В начале 80-х годов ХХ века, еще до того времени как стала известна молекулярная природа заболевания было предложено ввести термины "Болезнь Ниманна-Пика I типа" и "болезнь Ниманна-Пика II типа", которые, соответственно, использовались для обозначения высокого и низкого уровня накопления сфингомиелина в организме. Два типа болезни Ниманна-Пика, А и В возникают вследствие мутаций гена SMPD1, в то время как болезнь Ниманна-Пика типа С, вызывают мутации, происходящие в генах NPC1 и NPC2. Ранее, был еще один тип болезни - тип D, который использовался для того, чтобы отделить тех больных, которые происходили из Новой Шотландии. Ведь у этих лиц заболевания возникает из-за мутации гена NPC1. Однако, на сегодня такое разделение считают нецелесообразным и тип С включает в себя обе группы мутаций. В начале 80-х годов ХХ века, еще до того времени как стала известна молекулярная природа заболевания было предложено ввести термины "Болезнь Ниманна-Пика I типа" и "болезнь Ниманна-Пика II типа", которые, соответственно, использовались для обозначения высокого и низкого уровня накопления сфингомиелина в организме.мутацийгенахмутацийгенах
7 Болезнь Ниманна-Пика наследуется по аутосомно-рецессивному типу, т.е. для того, чтобы ребенок унаследовал это заболевание обе копии или аллели гена, унаследованные от родителей - должны быть мутировавшие (изменены, но так, что функции гена нарушаются, в отличие от полиморфизма, в котором нуклеотидная последовательность меняется, не вызывая при этом никаких функциональных нарушений). При этом, родители больного ребенка, зачастую являются носителями заболевания, и никаких признаков или симптомов болезни у них не проявляется. Если оба родителя - носители БНП, то вероятность того, что ребенок родится больным, составляет 25%. Именно поэтому, для тех семей, где известны случаи заболевания, необходимо осуществить генетическое тестирование и обратиться за генетической консультацией. Болезнь Ниманна-Пика наследуется по аутосомно-рецессивному типу, т.е. для того, чтобы ребенок унаследовал это заболевание обе копии или аллели гена, унаследованные от родителей - должны быть мутировавшие (изменены, но так, что функции гена нарушаются, в отличие от полиморфизма, в котором нуклеотидная последовательность меняется, не вызывая при этом никаких функциональных нарушений). При этом, родители больного ребенка, зачастую являются носителями заболевания, и никаких признаков или симптомов болезни у них не проявляется. Если оба родителя - носители БНП, то вероятность того, что ребенок родится больным, составляет 25%. Именно поэтому, для тех семей, где известны случаи заболевания, необходимо осуществить генетическое тестирование и обратиться за генетической консультацией. аутосомно-рецессивному типу,аллелиполиморфизмагенетическое тестированиегенетической консультацией. аутосомно-рецессивному типу,аллелиполиморфизмагенетическое тестированиегенетической консультацией.
13 Диагностика Некоторые формы болезни Ниманна - Пика можно диагностировать у плода на основании результатов исследования кусочка плаценты или амниоцентеза - пробы околоплодной жидкости, полученной путем пункции плодного пузыря. После рождения диагноз может быть подтвержден биопсией печени (берут кусочек ткани печени и исследуют под микроскопом). Некоторые формы болезни Ниманна - Пика можно диагностировать у плода на основании результатов исследования кусочка плаценты или амниоцентеза - пробы околоплодной жидкости, полученной путем пункции плодного пузыря. После рождения диагноз может быть подтвержден биопсией печени (берут кусочек ткани печени и исследуют под микроскопом).
14 Сочетание прогрессирующего снижения психических функций с гепатоспленомегалией и анемией должно настораживать в отношении болезни Нимана-Пика. Диагноз подтверждается биопсией костного мозга, периферических нервов или лимфатических узлов. При исследовании крови больных выявляются гипохромная анемия, тромбоцитопения. В периферической крови могут обнаруживаться вакуолизированные лимфоциты. Содержание свободного холестерина в крови повышено, иногда обнаруживается увеличениеконцентраций лецитина и сфингомиелина. Рентгенологически в костях обнаруживаются признаки остеопороза и остеомаляции. Сочетание прогрессирующего снижения психических функций с гепатоспленомегалией и анемией должно настораживать в отношении болезни Нимана-Пика. Диагноз подтверждается биопсией костного мозга, периферических нервов или лимфатических узлов. При исследовании крови больных выявляются гипохромная анемия, тромбоцитопения. В периферической крови могут обнаруживаться вакуолизированные лимфоциты. Содержание свободного холестерина в крови повышено, иногда обнаруживается увеличениеконцентраций лецитина и сфингомиелина. Рентгенологически в костях обнаруживаются признаки остеопороза и остеомаляции.
16 Лечение Возможности лечения болезни Ниманна-Пика в основном ограничены и преимущественно применяется поддерживающая и симптоматическая терапия. Стоит отметить, что операции по трансплантации органов, пока не очень успешны. В будущем ученые рассчитывают на то, что для лечения этого заболевания можно будет применять технологии ферментной замены и генную терапию. Возможности лечения болезни Ниманна-Пика в основном ограничены и преимущественно применяется поддерживающая и симптоматическая терапия. Стоит отметить, что операции по трансплантации органов, пока не очень успешны. В будущем ученые рассчитывают на то, что для лечения этого заболевания можно будет применять технологии ферментной замены и генную терапию.
17 При заболевании на тип В, может быть осуществлена трансплантация костного мозга. Однако, для улучшения качества жизни необходима поддерживающая терапия, которая включает контроль над питанием, постоянное употребление лекарств, надзор врачей и физиотерапию. При заболевании на тип В, может быть осуществлена трансплантация костного мозга. Однако, для улучшения качества жизни необходима поддерживающая терапия, которая включает контроль над питанием, постоянное употребление лекарств, надзор врачей и физиотерапию.
18 Дифференциальная диагностика Болезнь Ниманна Пика отличается от болезни Тея Сакса наличием спленомегалии, снижением активности сфингомиелиназы в лимфоцитах и, наконец, обнаружением клеток НиманнаПика в материале, полученном путем пункции грудины или селезенки. При дифференциации болезни Ниманна Пика от болезни Гоше необходимо иметь в виду, что при болезни Гоше, наряду с неврологическими симптомами и спленомегалией, выявляются иммунодефицитные состояния, обусловленные поражением костного мозга, а в фибробластах кожи и в клетках костного мозга обнаруживаются клетки Гоше. Болезнь Ниманна Пика отличается от болезни Тея Сакса наличием спленомегалии, снижением активности сфингомиелиназы в лимфоцитах и, наконец, обнаружением клеток НиманнаПика в материале, полученном путем пункции грудины или селезенки. При дифференциации болезни Ниманна Пика от болезни Гоше необходимо иметь в виду, что при болезни Гоше, наряду с неврологическими симптомами и спленомегалией, выявляются иммунодефицитные состояния, обусловленные поражением костного мозга, а в фибробластах кожи и в клетках костного мозга обнаруживаются клетки Гоше.
19 Прогноз При заболевании на тип А болезни Ниманна-Пика большинство пациентов умирают в возрасте до 18 месяцев. Относительно типов В и С, то прогноз развития в этих случаях более благоприятный. Обычно, пораженные этими типами люди живут до подросткового или зрелого возраста. При заболевании на тип А болезни Ниманна-Пика большинство пациентов умирают в возрасте до 18 месяцев. Относительно типов В и С, то прогноз развития в этих случаях более благоприятный. Обычно, пораженные этими типами люди живут до подросткового или зрелого возраста.
20 Профилактика Профилактика болезни Ниманна - Пика заключается в проведении медико-генетического консультирования и обследования в специализированных клиниках с целью выявления гетерозигот по аутосомно-рецессивному гену. Проводится также пренатальная диагностика: определяют активность сфингомиелиназы в культуре амниотических клеток. Профилактика болезни Ниманна - Пика заключается в проведении медико-генетического консультирования и обследования в специализированных клиниках с целью выявления гетерозигот по аутосомно-рецессивному гену. Проводится также пренатальная диагностика: определяют активность сфингомиелиназы в культуре амниотических клеток.
Болезнь Нимана-Пика. Разработка для студентов ВУЗов.

Болезнь Ниманна – Пика
Выполнила студентка 5 курса
Горюнова Яна Геннадьевна
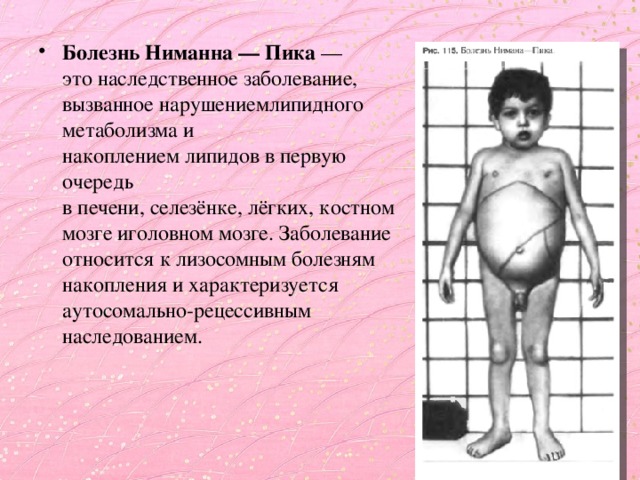
- Болезнь Ниманна — Пика — это наследственное заболевание, вызванное нарушениемлипидного метаболизма и накоплением липидов в первую очередь в печени, селезёнке, лёгких, костном мозге иголовном мозге. Заболевание относится к лизосомным болезням накопления и характеризуется аутосомально-рецессивным наследованием.
Различают три типа заболевания: типы A, B и C
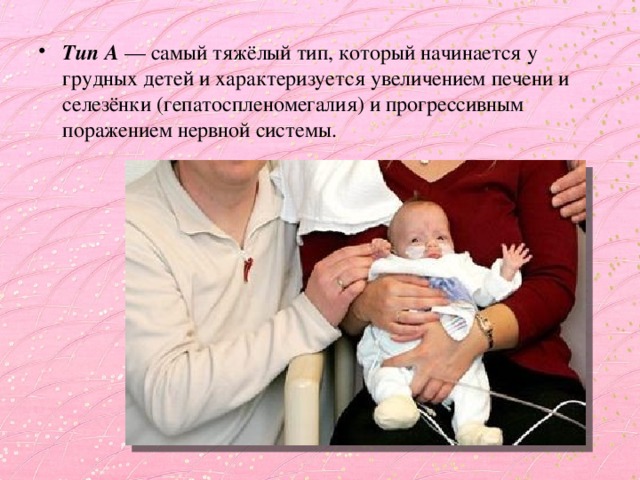
- Тип А — самый тяжёлый тип, который начинается у грудных детей и характеризуется увеличением печени и селезёнки (гепатоспленомегалия) и прогрессивным поражением нервной системы.

- Более умеренный тип B включает гепатоспленомегалию, задержку роста и нарушение лёгочной функции с частыми лёгочными инфекциями. Другие показатели включают повышенный уровень холестерина и липидов в крови, низкий счёт тромбоцитов (тромбоцитопения). Больные как правило доживают до взрослого возраста.

- Типы А и В вызываются мутациями гена кислой лизосомальной сфингомиелиназы ( SMPD1 ). Этот фермент отвечает за расщепление сфингомиелина в мембранах лизосом. Его недостаточность приводит к избыточному накоплению сфингомиелина, а как следствие и к более широкому нарушению липидного метаболизма, включая накопление холестерина и других липидов клетки.
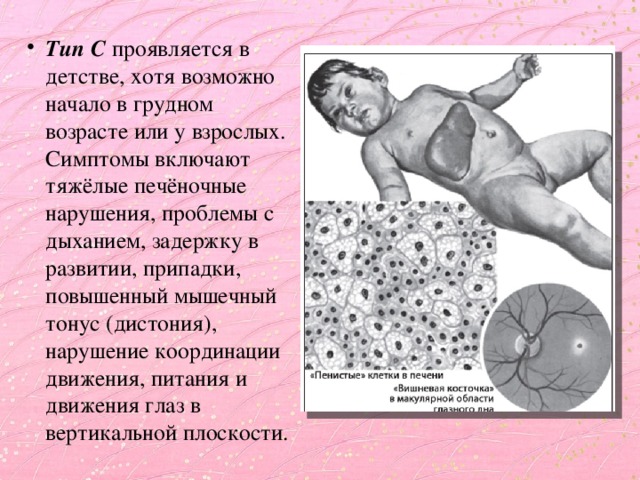
- Тип С проявляется в детстве, хотя возможно начало в грудном возрасте или у взрослых. Симптомы включают тяжёлые печёночные нарушения, проблемы с дыханием, задержку в развитии, припадки, повышенный мышечный тонус (дистония), нарушение координации движения, питания и движения глаз в вертикальной плоскости.

- Больные доживают до взрослого возраста. Частота заболевания — 1 на 150 тысяч.
- Этот тип болезни Ниманна — Пика вызывается мутациями генов NPC1 или NPC2 , которые кодируют белок клеточной мембраны, отвечающий за транспорт холестерина и липидов внутри клетки .
Лечение Болезни Нимана - Пика
- Лечение симптоматическое. Некоторая стабилизация процесса и улучшение общего состояния наблюдаются при назначении витаминов, переливании крови, плазмы, введении тканевых экстрактов.

2. Болезнь альцгеймера.
Крымский государственный медицинский университет
им. С.И. Георгиевского
АТРОФИЧЕСКИЕ ЗАБОЛЕВАНИЯ ГОЛОВНОГО
Фариков С.Э 406гр 1мед.
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА.
это неизлечимое нейродегенеративное
прогрессирующим снижением когнитивных
функций, в первую очередь, памяти, и
развитием поведенческих расстройств.
впервые описал заболевание в 1906 году

ИЗВЕСТНЫЕ ЛЮДИ С БОЛЕЗНЬЮ
2. Введение
▪ Болезнь Ниманна-Пика ведёт к
накоплению сфингомиэлина в
макрофагах селезенки, печени,
лимфатических желез, костного мозга,
легких, центральной нервной системы
и других тканей. Способ передачи
Этиология заключается в генетических

нарушениях. Болезнь появляется
исключительно у детей, обычно
грудных. Это редкое заболевание, оно
передается по наследству.
ВВЕДЕНИЕ
Болезнь Ниманна–Пика, тип С (НПС) — редкое прогрессирующее
наследственное аутосомно-рецессивное заболевание из группы
лизосомных болезней накопления. Патогенетические механизмы до
настоящего времени до конца не выяснены.
нарушениях. Болезнь появляется почти
4. Симптомы заболевания, типы.
▪ Клинические проявления ▪ Тип В-особенностью этого типа является отсутствие накопления липиднов в нервных
зависят от типа
клетках. Таким образом, нервные клетки не разрушаются и не появляются симптомы
заболевания. Для разных
нарушения деятельности головного мозга. У таких деток не страдают
типов характерны свои

интеллектуальные способности. Первые симптомы появляются после 3х лет. В первую
очередь начинает увеличиваться селезенка, позже печень. С возрастом появляются
особенности и тяжесть
симптомы поражения лёгких.
Тип А – ранее начало
развития болезни (3-5
-увеличение объёма живота

-нарушение работы печени и желудочного пузыря
-снижение массы тела
-одышка при умеренных физических нагрузках
-частые инфекции и простуда
Тип С- тип С проявляется после первых лет жизни. В начале происходит поражение

внутренних органов – увеличение печени и селезенки лимфатичких узлов.
-увеличение и болезненность лимфатических узлов
-частые бронхиты и воспаления лёгких
С течением болезни появляются симптомы поражения нервной системы, нарушается
работа головного и спинного мозга, больные отстают в психическом и физическом
развитии. Дети теряют навыки и умения, которыми уже овладели.
зависят от типа
Тип С- проявляется после первых лет жизни. В начале происходит поражение
4. факторы риска
ФАКТОРЫ РИСКА
наследственность — наличие в семье случая заболевания
неконтролируемая артериальная гипертензия (повышение
атеросклероз артерий, кровоснабжающих головной мозг;
черепно-мозговая травма в анамнезе;
депрессия в молодом возрасте;
женский пол в связи с большей продолжительностью жизни;
низкий уровень образования и низкая интеллектуальная
7. Патогенез
ЭТИОПАТОГЕНЕЗ
выявлены 4 гена (пресенелин 1,
пресенелин 2, ген, кодирующий
белка, ген, кодирующий
аполипопротеин Е-4), носительство
которых связано с высоким риском
ЭТИОПАТОГЕНЕЗ
атрофия обусловлена генетически и
▪ патогенез Ниманна – Пика был связан с недостаточностью в
тканях сфингомиелиназы- кислота лизосомах гидролазы,
осуществляющие гидролитическое расщепление
сфингомиелина. При недостаточности сфингомиелиназы
нарушается нормальный катаболизм сфингомиелина и
происходит накопление его в тканях.
Возможность избыточного синтеза сфингомиелина при
Ниманне- Пике болезнь экспериментально не подтвердилась,
но полностью пока не исключена.
Ниманна- Пика болезнь экспериментально не подтвердилась,
но полностью пока не исключена
ЭПИДЕМИОЛОГИЯ, ГЕНЕТИКА, ОСНОВНЫЕ МЕХАНИЗМЫ
• НПС относится к редким наследственным болезням
обмена веществ с суммарной распространенностью в
популяции 1:130 000 живых новорожденных.
• Болезнь НПС обусловлена мутациями генов NPC1 или
NPC2. Молекулярные механизмы развития болезни НПС
пока еще полностью не расшифрованы.
• При микроскопическом исследовании во всех органах
обнаруживают пенистые клетки, или т.н. клетки
1. Болезнь Нимана-Пика
▪ К сожалению, прогнозы для
пациентов с подобным диагнозом
неутешительные. Болезнь Пика очень серьёзная, на сегодняшний
день не существует способов
остановить разрушение нейронов.
Согласно статистике, в течение 510лет болезнь приводит к
психическому и моральному
разложению личности – у
слабоумие, маразм и так далее.
Такие больные должны находится
под постоянным присмотром,
желательно в специальных
клиниках, где за ними будут
постоянно следить медицинские
Согласно статистике, в течение 510 лет болезнь приводит к
Выполнила: студентка 5 курса
5 группы, 4 мед. Ф-та
При заболевании на тип А болезни
пациентов умирают в возрасте до
18 месяцев. Относительно типов В
и С, то прогноз развития в этих
пораженные этими типами люди
живут до подросткового или
2. Болезнь альцгеймера.
АЛОИС АЛЬЦГЕЙМЕР
11 фотографиях. В
11 фотографиях. В
7. образование ß-амилоида
ОБРАЗОВАНИЕ ß-АМИЛОИДА
участки, один из
ключевую роль в
12. Возможно, это прозвучит ужасный, но члены семьи открыто говорят, что молится о приближении конца.
▪ Иногда она улыбается и
даже смеется. Не
понимаю, по какой
причине. Мы надеемся
что развязка близка” говорил сын.
даже смеется. Не
понимаю, по какой
9. патоморфология
Паталогоанатомическое исследование обнаруживает
значительное увеличение размеров и желтую окраску
печени и селезенки, пятнистый рисунок легких.
Надпочечники также значительно увеличены в
размерах и содержат большое количество липидов.
При микроскопии во всех органах обнаруживаются
клетки, которые при фиксации спиртом выглядит
достигать значительной величины 20-25мкм, в
некоторых случаях 90 мкм. Пенистость клеток- это
артефакт, вызванный рстворением жироподобных
субстанций, содержащихся в клетках. Клетки НиманаПика могут быть онаружены при жизни в пунктатах
селезнки, костного мозга. Крупные клетки с пенистой
протоплазмойне специфичны для болезни НиманаПика и могут обнаруживаться при других липидозах и
гиперлипидемии. Однако они хорошо отличаются от
ПАТОМОРФОЛОГИЯ
(PiD) is one of the frontal
by the presence of
(silver staining) spherical
inclusions called Pick
bodies and globose
While Pick bodies are primarily found in
neurons, small, Pick-body like inclusions
have been described in glial cells
14. Заключение
ЗАКЛЮЧЕНИЕ
Болезнь НПС требует выполнения тщательной дифференциальной диагностики с большим
числом и наследственных, и ненаследственных заболеваний.
Прогноз заболевания независимо от возраста манифестации НПС неутешителен.
На сегодняшний день уже существует возможность не только приостановить
прогрессирование болезни, но и достигнуть обратного развития отдельных ее клинических
проявлений и улучшить качество жизни пациента. В связи с этим трудно переоценить
значимость ранней диагностики НПС, необходимой для своевременного назначения
▪ Полностью излечить заболевание врачам
пока не удается, но правильно подобранная
терапия может существенно снизить
тяжесть симптомов и улучшить качество
10. Клиника
В начале болезни наблюдается отказ ребенка
отпищи, периодическая рвота; очень рано
увеличиваются размеры печени и селезенки ,
развивается гипотрофия. Появление признаов,
указывающих на поражение нервной системы,
также позволяет заподозрить болезнь НиманаПика. Спастические парезы могут сменяться
общей мышечной гипотонией, гипорефлексией.
Прогрессирующее поражение нервной системы
ведет к резкому отставанию ребенка в нервнопсихическом развитии, появлению глухоты,
слепоты. У 20-30% детей при осмотре глазного
дна обнаруживается симптом «вишнёвой
коричневатый оттенок. Резистентность к
инфекции снижены: дети подвержены
заболеваниям легких (пневмонии), ушей (отиты).
Общими для всех форм симптомами являются
увеличение лимфатических узлов, Обычно
отмечаются побочные признаки гиперспленизма.
Характерна инфильтрация лёгких, выявляемая
рентгенологически. Неврологическая симптоматика
(отсутствующие при висцеральной форме
заболевания , тип В)включают задержку
психомоторного развития, атаксию, судороги,
снижение мышечного тонуса и угнетение сухожльных
рефлексов. У некоторых больных при исследовании
глазного дна обнаруживают симптом «вишневой
нодулярные ксантомы на коже. В периферической
крови, чаще в костном мозге, а также в печени,
селезенке, почках, надпочечниках, лимфатических
узлах и некотрых других органах обнаруживаются
довольно крупные зернистые и вакуолизированные «
при болезни Нимана-Пика обусловлены инактивацией
энзима сфингомиелиназы, что приводит к нарушению
катаболизма сфингомиелина и накоплению его в
клетках пораженных органов.
КЛИНИКА
болеет 5% лиц старше 65 лет
причина слабоумия в 30-40% случаев
продолжительность от 2 до 10 лет
смерть наступает из-за сопутствующих
заболеваний или осложнений, возникших изза отсутствия движений
КЛИНИКА. ПЕРВАЯ СТАДИЯ
симптомы нарушения социального
симптомы немотивированных поступков
заострение эгоистической ориентации
симптом грамафонной пластинки
мория или апатия
11. Инициальный период
ИНИЦИАЛЬНЫЙ ПЕРИОД
затяжные невротические состояния
затяжные депрессивнные эпизоды
хронические параноидные состояния
идеи ревности и ущерба
снижение успешности социального
29. вторая стадия
ПЕРВАЯ СТАДИЯ – МАНИФЕСТНАЯ
прогрессирующее расстройство памяти
реакция личности на когнитивный дефицит
амнезия на привычные действия
ВТОРАЯ СТАДИЯ
мышечная ригидность, паркинсонические проявления,
персеверации в речи
полное отсутствие критики
кратковременные эпизоды психозов
апраксия
ТРЕТЬЯ СТАДИЯ – МАРАНТИЧЕСКАЯ
мышечный тонус повышен
переход в вегетативную кому
мория или апатия
ВТОРАЯ СТАДИЯ
очаговые симптомы в форме амнезии,
афазии, апраксии, агнозии, акалькулии
гиперальгезия кожных покровов
маразм с переходом в вегетативную кому
18. диагностика. критерии диагностики.
ДИАГНОСТИКА. КРИТЕРИИ ДИАГНОСТИКИ.
наличие критериев деменции
постепенное начало с медленно
отсутствие данных в пользу др. заболевания
отсутствие очаговой мозговой симптоматики
на ранних этапах деменции
PET-сканирование
мозга при болезни
угасание активности в
кое скринингтестирование, при
20. лечение. фармакотерапия
ЛЕЧЕНИЕ. ФАРМАКОТЕРАПИЯ
борьба с когнитивным дефицитом
ингибиторы холинэстеразы (физостигмин,
мегавитаминная терапия (В5 В12,В2, Е)
ЛС, влияющие на мозговой кровоток (кавинтон)
ЛС, влияющие на процессы памяти (соматотропин, префизон,
антипсихотики в малых дозах в периоды острого психоза
антагонист глутаматных рецепторов( мемантин, уменьшает
воздействие глутамата — основного возбуждающего медиатора в
нервной системе — на нейроны и предотвращает их гибель –
шунтирование против ликворной гипертензии
21. Психосоциальное вмешательство
ПСИХОСОЦИАЛЬНОЕ ВМЕШАТЕЛЬСТВО
Психосоциальное вмешательство дополняет фармакологическое и может
Что такое болезнь Ниманна-Пика?
Болезнь Ниманна-Пика (БПН, дефицит кислой сфингомиелиназы) представляет собой редкое прогрессирующее генетическое заболевание, относящиеся к группе лизосомных болезней накопления, которое возникает в результате дефицита ферментной кислой сфингомиелиназы, которая необходима для расщепления (метаболизма) жирного вещества (липида), называемого сфингомиелином. Следовательно, сфингомиелин и другие вещества накапливаются в различных тканях организма (печень, селезенка, головной мозг, костный мозг).
Болезнь Ниманна-Пика сильно изменчива, возраст возникновения, конкретные симптомы и степень тяжести расстройства могут значительно варьироваться от одного человека к другому, иногда даже среди членов одной семьи.
Это расстройство лучше всего рассматривать как спектр заболеваний. В тяжелом конце спектра находится фатальное нейродегенеративное расстройство, которое проявляется в младенчестве (болезнь Нимана-Пика типа А [БНПА]). На мягком конце спектра, больные люди не имеют или имеют только минимальные неврологические симптомы, и часто встречается больные доживающие до взрослого возраста (болезнь Нимана-Пика, тип B [БНПВ]). Существуют также промежуточные формы расстройства. БПН вызывается мутациями в Ген SMPD1 и наследуется по аутосомно-рецессивному типу.
Классификация
Существуют три расстройства, известные как болезнь Ниманна-Пика, типы A, B и C. Эти расстройства были изначально сгруппированы из-за схожих симптомов, но теперь стало известно, что это разные заболевания. Типы БНП A и B обусловлены мутациями в гене SMPD1, которые вызывают дефицит специфического фермента, кислой сфингомиелиназы. Болезнь Ниманна-Пика типа C обусловлен мутациями в одном из двух разных генов и не содержит дефицитного фермента. По заявлению NORD БНП типа C в настоящее время считается отдельным заболеванием, отличительным от БНП типа A и B.
БНП традиционно делится на две подгруппы:
- нейронопатические (тип A);
- ненейропатические (тип B).
Нейронопатическая относится к расстройствам, которые повреждают клетки мозга (нейроны). Тип A обычно вызывает серьезные нейродегенеративные заболевания в младенческом возрасте, в то время как тип B обычно не считается неврологическим заболеванием. Однако, поскольку случаи заболевания находятся между этими двумя крайностями, такие широкие обозначения могут вводить в заблуждение людей. Некоторые исследователи используют болезнь типа B для того чтобы сослаться на все мягкие и промежуточные формы заболевания, которые могут иметь неврологические результаты.
Признаки и симптомы болезни Ниманна-Пика
Поскольку Болезнь Ниманна-Пика является сильно изменчивым расстройством, важно отметить, что у затронутых лиц не будет всех симптомов, описанных ниже, и что каждый отдельный случай уникален. У некоторых детей развиваются тяжелые, угрожающие жизни осложнения в начале жизни; у других — легкое заболевание, которое может не диагностироваться до взрослого возраста. Родители должны поговорить с врачом своего ребенка о конкретных симптомах и общем прогнозе.

Тяжелую, инфантильную форму БНП, известную как тип А, можно отличить от более легких форм, которые появляются позже. Первичным симптомом в большинстве детских случаев является аномальное увеличение печени и/или селезенки (гепатоспленомегалия), которое может прогрессивно ухудшаться, пока печень и селезенка не станут массивными. Также может происходить значительное накопление жидкости в брюшной полости (асцит). В период новорожденности могут возникнуть пожелтение кожи и склеры глаз (желтуха).
Дополнительные симптомы в младенческом возрасте включают:
- проблемы с питанием;
- запоры;
- тошноту;
- рвоту;
- желудочно-кишечный рефлюкс;
- раздражительность;
- потерю рефлексов;
- прогрессирующую потерю мышечного тонуса (гипотония).
Проблемы с питанием и другие отклонения (например, частая рвота) могут привести к неспособности к здоровому развитию.
Накопление сфингомиелина в легких может привести к рецидивирующим респираторным инфекциям и затруднению дыхания, что потенциально может привести к угрожающей жизни легочной недостаточности.
У большинства младенцев развивается состояние, известное как пятна Пика (вишнево-красные пятна на глазах). Вишнёво-красные пятна поражают макулу — область сетчатки, содержащую клетки, чувствительные к свету, необходимые для центрального зрения. Обычно оно желтое. Пятна Пика не всегда присутствует у поражённых людей.
Достижение основных этапов развития и общего развития может быть нормальным в первые несколько месяцев. Однако часто в возрасте от 3 до 12 месяцев развитие осложняется, и пораженные дети теряют ранее приобретенные двигательные навыки. Пострадавшие дети могут испытывать глубокие неврологические ухудшения, повышение мышечного тонуса и ригидности мышц (спастичность), а к 3 годам это заболевание часто приводит к летальному исходу.

У людей с поздними формами БНП могут развиться симптомы как в младенчестве, так и во взрослой жизни. Иногда эти формы все вместе называют болезнью Ниманна-Пика типа В; они, как правило, слабее, чем БНП типа А (инфантильная форма). Люди с легкими формами могут дожить до поздней взрослой жизни, а некоторые могут оставаться недиагностированными до взрослого возраста. Болезнь Немана-Пика типа В ассоциируется с системным заболеванием, которое может сильно различаться по степени тяжести и распространенности.
Гепатоспленомегалия является распространенным начальным признаком и может варьироваться от легкого до серьезного увеличения органов. Прогрессирующее увеличение селезенки может вызвать низкий уровень тромбоцитов и лейкоцитов. Белые кровяные клетки (лейкоциты) помогают бороться с инфекцией, и уменьшенное количество этих клеток может сделать пораженного человека восприимчивым к инфекции. Тромбоциты — это специализированные клетки крови, которые позволяют организму образовывать сгустки и остановить кровотечение. Уменьшенное количество тромбоцитов, известное как тромбоцитопения, может привести к эпизодам длительного кровотечения.
Результатом увеличения печени и селезенки может быть боль в животе. Увеличенная селезенка подвержена риску разрыва, что может привести к опасному для жизни кровотечению в брюшную полость.
У большинства людей с болезнью типа В присутствует определенная степень заболевания печени. Большинство из них имеют аномальные анализы крови печени и рубцы в печени. Рубцевание может варьировать от легкого без симптомов до откровенного цирроза и печеночной недостаточности.
У некоторых пострадавших наблюдается постепенное ухудшение функции легких. Для некоторых людей поражение легких может быть легким без заметных симптомов. У некоторых людей может возникнуть затруднение дыхания при физической нагрузке (одышка). Другие люди могут испытывать постоянное ухудшение дыхательной (легочной) функции с серьезными ограничениями в уровнях активности и кислородной зависимости. Может возникнуть рецидивирующая пневмония.
У людей с поздним детским БНП обычно не развиваются неврологические симптомы, но могут развиваться легкие симптомы, или, в редких случаях, могут развиваться клинически значимые неврологические симптомы. У некоторых пострадавших детей и подростков могут развиться:
- быстрые непроизвольные движения глаз (нистагм);
- можечковые нарушения, которые включают неустойчивую манеру ходьбы и неуклюжесть.
Интеллектуальной инвалидности и психических расстройств также нет. Могут возникнуть:
- аномалии сетчатки;
- аномалии богатой нервами мембраны, выстилающей заднюю часть глаз;
- периферическая нейропатия.
Периферическая нейропатия является общим термином для любого заболевания, поражающего нервы вне центральной нервной системы. Общие симптомы включают потеря чувствительности или возникновение неприятных ощущений, таких как зуд, жжение или покалывание вдоль пораженных нервов.
У большинства затронутых детей наблюдаются задержки в росте и малый вес, хотя большинство в конечном итоге достигает почти нормального роста к взрослой жизни. Задержка полового и скелетного созревания также могут возникнуть. У большинства людей присутствует остеопения (истончение костей).
Пострадавшие люди могут быть подвержены риску заболевания коронарных артерий на ранних стадиях.
Причины болезни Ниманна-Пика

Болезнь Ниманна-Пика обусловлена мутацией в гене сфингомиелин фосфодиэстеразы-1 (SMPD1). Гены предоставляют инструкции для создания белков, которые играют критическую роль во многих функциях организма. Когда происходит мутация гена, белковый продукт может быть дефектным, неэффективным или отсутствовать. В зависимости от функций конкретного белка это может повлиять на многие системы органов организма, включая мозг.
Ген SMPD1 создает (кодирует) фермент, известный как кислая сфингомиелиназа. Мутация в этом гене приводит к недостаточным уровням функциональных копий фермента кислой сфингомиелиназы. Этот фермент необходим для расщепления (метаболизма) определенных жирных веществ (липидов) в организме. Снижение или отсутствие активности фермента приводит к аномальному накоплению сфингомиелина в различных тканях организма. Сфингомиелин — это жирное вещество, являющееся компонентом большинства клеточных мембран. Ненормальное накопление сфингомиелина в определенных тканях организма вызывает признаки и симптомы болезни Ниманна-Пика.
Болезнь Ниманна-Пика — это генетическое заболевание, которое передается по наследству от родителей и может присутствовать у других членов семьи. В целом, люди получают две копии большинства генов, один наследуется от матери, а другой от отца. Рецессивные генетические нарушения, такие как болезнь Ниманна-Пика, возникают, когда человек наследует изменения или мутации в обеих копиях определенного гена (в этом случае SMPD1 ген.).
Если человек получает одну нормальную копию гена и одну измененную копию, человек будет носителем болезни, но не проявлять признаков болезни. Риск для двух родителей-носителей, передать дефектную копию гена и, следовательно, зачать больного ребенка, составляет 25% при каждой беременности. Риск родить ребенка, который будет носителем, как родители, составляет 50% для каждой беременности. Шанс для ребенка получить нормальные копии гена от обоих родителей и не быть ни больным, ни носителем этой конкретной мутации составляет 25%. Риск одинаков как для мужчин, так и женщин.
БНП поражает мужчин и женщин в равных количествах. Точная частота и распространенность этого заболевания неизвестны, но оценивается 1 к 250 000 человек в общей популяции. Однако из-за того, что некоторые случаи неправильно диагностируются или не диагностируются вообще, определить истинную частоту болезни в общей популяции сложно. Тяжелая инфантильная форма (тип А) может поражать различные этнические группы, но встречается с большей частотой у лиц еврейского происхождения. Более поздние формы (тип B) могут затрагивать все этнические группы.
Схожие расстройства
Симптомы следующих расстройств могут быть похожи на симптомы БНП. Сравнения могут быть полезны для дифференциальной диагностики.
Существует несколько типов метаболических нарушений, при которых происходит вторичное накопление определенных веществ, таких как жиры и углеводы, включая мукополисахаридозы (группа метаболических заболеваний соединительной ткани) и другие лизосомные болезни накопления. Эти расстройства включают:
- галактоземию;
- галактосиалидоз;
- сиалидоз;
- болезнь Ниманна-Пика типа C;
- болезнь Гоше;
- болезнь Вольмана.
Диагностика
Диагноз заболевания основан на выявлении характерных признаков, детальной истории болезни, тщательной клинической оценке и разнообразных специализированных тестах.
Лечение болезни Ниманна-Пика
Лечение болезни Ниманна-Пика может потребовать скоординированных усилий команды специалистов. Педиатрам, неврологам, гепатологам, офтальмологам и другим медицинским работникам может потребоваться систематическое и всестороннее планирование аффективного лечения ребенка. Психосоциальная поддержка для всей семьи также важна. Генетическое консультирование может быть полезным для пострадавших людей и их семей.
Современные методы лечения направлены на конкретные симптомы, которые проявляются у каждого человека. В случае БНП типа A может быть рекомендована физическая и профессиональная терапия и периодическая оценка питания. Для обеспечения правильного питания может потребоваться имплантация питательной (гастрономической) трубки. При этой процедуре тонкая трубка помещается в желудок через небольшой разрез в брюшной полости, что позволяет непосредственно принимать пищу и/или лекарства. Проблемы со сном, связанные с расстройством, можно лечить с помощью ночных успокоительных средств.
Некоторым взрослым с болезнью типа В может потребоваться лечение дислипидемии (изменение уровней холестерина и/или триглицеридов). Питательная поддержка также рекомендуется для людей с заболеванием типа B, чтобы обеспечить достаточное количество калорий, правильное потребление кальция и витамина D из-за риска остеопении и снизить риск дислипидемии у взрослых.
Пациентам с болезнью Ниманна-Пика типа B, у которых длительное кровотечение вызвано тромбоцитопенией, может потребоваться переливание крови. Лицам с заболеваниями легких может потребоваться вспомогательная кислородная терапия. Лицам с увеличенной селезенкой рекомендуется избегать контактных занятий спортом во избежание разрыва селезенки. Взрослые с гиперлипидемией должны контролировать уровень холестерина. Также сообщалось о случаях трансплантации печени у некоторых взрослых с печеночной недостаточностью, вызванной БНП типа B.
Прогноз
Болезнь Ниманна-Пика типа А является смертельной в раннем детстве. Клиническое проявление и течение БНП типа A является относительно однородным и характеризуется нормальностью ребенка при рождении, сопровождаемый прогрессирующей гепатоспленомегалией с 3-х месяцев и тяжелым нейродегенеративным течением, приводящим к смерти к 3 годам.
В отличие от стереотипного фенотипа типа А, клиническая картина и течение пациентов с заболеванием БНП типа В более вариабельны в отношении клинических данных, возраста начала и тяжести симптомов. Большинство пациентов диагностируется в младенчестве или детстве, когда во время обычного физического обследования обнаруживается увеличение печени и селезенки. С данным типа заболевания дети доживают до зрелого возраста.
Ребенок с признаками типа C до 1 года (хотя это др. заболевание) может не дожить до школьного возраста. Те, у кого появляются симптомы после поступления в школу, могут жить в подростковом возрасте. Некоторые могут жить в свои 20 лет.
Читайте также: