
Доброкачественный сонный миоклонус новорожденных что это
г. Симферополь
г. Симферополь
Режимы работы:
Телефоны
Приемное отделение
Колл-центр Справочной службы КДЦ( Консультативно-диагностический центр ):
Регистратура КДЦ:
Платные услуги:
Приемная главврача:
КДЦ( Консультативно-диагностический центр ):
- Главная
- Статьи
- НЕЭПИЛЕПТИЧЕСКИЕ СОСТОЯНИЯ У ДЕТЕЙ. ДОБРОКАЧЕСТВЕННЫЙ НЕЭПИЛЕПТИЧЕСКИЙ МИОКЛОНУС МЛАДЕНЧЕСТВА.
В настоящее время увеличивается количество судорожных состояний у детей. Чаще всего они провоцируются тяжелыми поражениями головного мозга, генетическими аномалиями, обменными нарушениями. Данные состояния требуют обязательного назначения противосудорожных препаратов и наблюдения детского невролога.
Наряду с этим, все чаще встречаются у детей приступы, вызывающие волнение родителей, но не нуждающихся в медикаментозной коррекции. Это неэпилептические приступы, связанные с неспецифическими реакциями незрелого мозга.
Синдром Феджермана – доброкачественные неэпилептические спазмы младенчества, внешне практически неотличимые от эпилептических приступов, таких как инфантильные судороги или миоклонические приступы.
Причина возникновения синдрома Феджермана неизвестна. По данным Maydell (2001), двигательные проявления синдрома представляют собой физиологические спазмы мышц. По сообщению Caraballo и соавт. (2009), интересной особенностью большинства обследованных семей пациентов с синдромом Феджермана является наличие хотя бы у одного из родителей университетского образования. Имеет ли это значение – покажут будущие исследования.
Прогноз синдрома Феджермана благоприятный с полным излечением к двух-трехлетнему возрасту (чаще приступы прекращаются на втором году жизни). Нарушений психоречевого развития не отмечается ни у одного пациента. Сочетание синдрома Феджермана с идиопатической фокальной эпилепсией (оба возрастзависимых состояния) может свидетельствовать о наличии единого механизма патогенеза – врожденного нарушения процессов созревания мозга.
Проявления – доброкачественного миоклонуса младенчества следует оличать в первую очередь, в первую очередь, от младенческих спазмов и эпилептических приступов, однако следует помнить и о других неэпилептических судорожных состояниях. Это дистонические феномены, характерные для пароксизмальных дискинезий, синдрома Сандифера, пароксизмальной кривошеи, пароксизмального хореоатетоза. Дрожание может наблюдаться при медикаментозных отравлениях, судорогах младенцев, а также в рамках доброкачественного тремора. Кивки и вздрагивания следует отличать от доброкачественных судорог новорожденного и других эпилептических приступов. С тоническими вытягиваниями могут быть сходны доброкачественный поворот глазных яблок, изменение позы при испуге и вздрагиваниях.
С учетом жалоб родителей, данных осмотра, нормального психомоторного развития, отсутствия в неврологическом статусе очаговой симптоматики, а также неизмененной возрастной ЭЭГ устанавливается диагноз: доброкачественный неэпилептический миоклонус младенчества. Рекомендовано воздержаться от назначения длительной противосудорожной терапии. Дальнейшее наблюдение за детьми показало постепенное исчезновение указанных состояний. При возникновении у ребенка насильственных вытягиваний, вздрагиваний, складываний, вычурных поз, поворотов головы и глаз – необходимо обратиться к врачу для своевременного определения причины данных состояний и решения вопроса о назначении лечения.

Аннотация научной статьи по клинической медицине, автор научной работы — Миронов М. Б., Ноговицын В. Ю., Абрамов М. О., Домбровская Е. А., Кваскова Н. Е.
Синдром Феджермана ( доброкачественный неэпилептический миоклонус младенчества ) -редкий тип пароксизмальных неэпилептических состояний, диагностируется на основании типичных клинических проявлений в виде кратковременных пароксизмальных кивков или вздрагиваний при условии отсутствия очаговой неврологической симптоматики и нормального психомоторного развития у детей в период младенчества, при этом не должны регистрироваться эпилептиформные изменения на ЭЭГ как в период пароксизма, так и интериктально. Синдром Феджермана возникает обычно на первом году жизни (чаще в 6 месяцев).Прогноз благоприятный с полной спонтанной ремиссией к двух-трехлетнему возрасту. В отечественной литературе синдром Феджермана представлен в единичных публикациях, в связи с чем приведены собственные клинические наблюдения.
Похожие темы научных работ по клинической медицине , автор научной работы — Миронов М. Б., Ноговицын В. Ю., Абрамов М. О., Домбровская Е. А., Кваскова Н. Е.
FEJERMAN SYNDROME (benign nonepileptic myoclonus of infancy)1St. Luka’s Institute of Child Neurology and Epilepsy
Fejerman syndrome ( benign nonepileptic myoclonus of infancy ) is a rare nonepileptic paroxysmal disorder, characterized by typical presentation of short head nodding or shuddering in otherwise healthy infant without focal signs or psychomotor retardation and not associated with epileptiform abnormalities on EEG. Onset of this disorder occurs at first year of life (predominantly 6 months). Prognosis is benign with spontaneous disappearing of paroxysmal episodes by age of 2-3 years. This syndrome is poorly described in Russian literature. We present the description of our own clinical cases.
Российская Противоэпилептическая Лига
и пароксизмальные состояния
Включен в перечень ведущих рецензируемых журналов и изданий ВАК
2013 Том 5 №2 и паР°кс^™ьлТ“;
СИНДРОМ ФЕДЖЕРМАНА (доброкачественный неэпилептический миоклонус младенчества)
Миронов М.Б.1, Ноговицын В.Ю.2, Абрамов М.О.1,
Домбровская Е.А.2, Кваскова Н.Е.1, Мухин К.Ю.1
1 Институт детской неврологии и эпилепсии имени Святителя Луки, Москва
2 Морозовская детская клиническая больница, Москва
Резюме: синдром Феджермана (доброкачественный неэпилептический миоклонус младенчества) -редкий тип пароксизмальных неэпилептических состояний, диагностируется на основании типичных клинических проявлений в виде кратковременных пароксизмальных кивков или вздрагиваний при условии отсутствия очаговой неврологической симптоматики и нормального психомоторного развития у детей в период младенчества, при этом не должны регистрироваться эпилептиформные изменения на ЭЭГкак в период пароксизма, так и интерик-тально. Синдром Феджермана возникает обычно на первом году жизни (чаще в 6 месяцев).Прогноз благоприятный с полной спонтанной ремиссией к двух-трехлетнему возрасту. В отечественной литературе синдром Феджермана представлен в единичных публикациях, в связи с чем приведены собственные клинические наблюдения.
Ключевые слова: синдром Феджермана, доброкачественный неэпилептический миоклонус младенчества, эпилептические спазмы, видео-ЭЭГ-мониторинг.
Синдром Феджермана диагностируется на основании типичных клинических проявлений в виде кратковременных пароксизмальных кивков или вздрагиваний при условии отсутствия очаговой неврологической симптоматики и нормального психомоторного развития у детей в период младенчества, при этом не должны регистрироваться эпилептиформные изменения на ЭЭГ как в период пароксизма, так и инте-риктально [4].
Доброкачественный неэпилептический миоклонус младенчества возникает обычно на первом году жизни (чаще в 6 мес.), совпадая с возрастом дебюта синдрома Веста [15]. В исследовании Caraballo и соавторов (2009) дебют ДНММ наблюдался в возрастном интервале от 1 до 12 мес. жизни, что в среднем соста-
Тел. (495) 983-09-03
вило 5,6 месяцев. Обычно пароксизмы кратковременны (1-2 сек.), но возможны и более длительные эпизоды обычно за счет возникновения кластеров и серий (в 40% случаев). Серийные и изолированные движения могут наблюдаться несколько раз в день, но необязательно ежедневно [4].
По данным Panayitopoulos (2010), моторные феномены в рамках ДНММ могут различаться по кинематике. Часть событий проявляется короткими тоническими спазмами конечностей или шеи, другие
В своей статье Caraballo и соавт. (2009) [4], проанализировав большую группу пациентов (n=102) с ДНММ, суммировали различные кинематические варианты данных пароксизмов (см. табл. 1). Принципиально все движения были разделены на миоклонус, дрожательные пароксизмы и спазмы с кратковременным тоническим напряжением конечностей и/ или головы и шеи.
Добрый день, просьба помочь разобраться в ситуации. Очень хотелось бы получить комментарий доктора Василий Юрьевича (drwn). Краткая история:
У нас мальчик - 4,5 мес. (род. 10.07.15).
Роды были обычными - плановое кесарево, по апгар 8 из 8. Беременность проходила нормально, за исключением временного повышения АСТ и АЛТ в середине беременности и с 7-8 месяца был зуд беременных (при этом была повышена только щелочная фосфатаза). При рождении образовалась небольшая субэпендимальная киста 4 мм в левой каудо-таламической вырезке (сказали это нормально у кесарят). Прививку делали только БЦЖ в роддоме. Малыш стал развиваться хорошо.
Месяц назад (где то в 3-3,5 мес.) заметили что сын плохо спит ночью - может по часу спать, просыпаться, потом опять засыпать (до этого спал и по 2-3 часа). Было ощущение, что ему что то мешает. Думали на колики.
18.11.15 начали делать ему общеукрепляющий массаж (до этого делали месяц назад - все ок) - не по показаниям, а просто ради доп. пользы. Возможно дальнейшее было спровоцировано массажем.
21-22 ноября 15 года вечером заметили, что сын непроизвольно в состоянии покоя стал делать движения, как в рефлексе Моро (см. видео). Дико перепугались. До этого рефлекс был только при испуге (и то нам его показывал невропатолог), а тут он сам по себе и несколько раз (в сериеи 1-10 приступов, по 5-7 серий в день). Массаж отменили (7 курсов из 10 сделали).
25.11.15 Посетили невролога, которая сделала повторное узи (киста уменьшилась до 3,5 мм), обследовала ребенка (заболеваний нервной системы нет, психомоторное развитие соответствует возрасту) и направила к неврологу-эпелептологу и на ЭЭГ. Сказала, что малыш хорошо развит.
25.11.15 Сделали ЭЭГ (правда малышу было постоянно неудобно, он капризничал после того как ему надели шапочку, даже потом остались сильные раздражения где были контакты, потом сказали, что хорошие детские шапочки для ЭЭГ были на сервисном обслуживании в тот день) - результат: "В ЭЭГ выявляются выраженные диффузные общемозговые изменения биоэлектрической активности в виде дизритмии, признаков раздражения стволовых структур, политопной эпилептиформной активности, сопровождающейся клиническими проявлениями.". Малыш несколько раз проявил судороги при ЭЭГ, но не всегда они отображались одновременно с разрядом. Доктор написал: "Изредка сразу за разрядом клинически отмечаются движения в виде вдрагивания, разведения рук".
26.11.15 Пошли к неврологу-эпелиптологу (сказали, что лучший в городе - к.м.н) - результат: по ЭЭГ она не видит эпи-активности, нужно пересдать (через 3 нед.). Мы честно говоря пропылесосив весь интернет подумали на синдром Веста, но врач сказала, что должна быть гипсаритмия, которой у нас по ЭЭГ нет. Так же должны быть инфантильные спазмы (которые якобы невозможно ни с чем перепутать и от них нельзя отвлечь/переключить ребенка во время приступа). У нас же малыш проявил спазмы при ней и она сказала, что это точно не они (и вообще это не имеет отношения к эпилепсии). Так же сказала, что у нас чудный малыш и он отлично развит по возрасту. На наш вопрос, что до проявления спазмов бывает, что младенцы развиваются нормально, она сказала, что такое практически невозможно и были бы по нему уже заметны проявления повреждений. Поставила диагноз "Доброкачественный миоклонус младенчества, психомоторное развитие соответствует возрасту." И что ничего нам принимать не нужно, но может быть только витамин Б-6.
На следующий день (27.11.15) вызвали на дом невролога, которая работает с ДЦП-шными детками: она нас несколько раз предупредила, что ее квалификация не имеет отношения к эпилепсиям, но она может определить при осмотре какая часть мозга может быть поражена/раздражена. Далее после 3-х признаков она сказала, что видит раздражение ствола и возможно это спазм сосудов/артерий ствола мозга. Признаки такие: 1) у нас есть пятно аиста/родимое на затылке (а оно появляется либо при стоячем положении плода при беременности, либо при травме-растяжении если плод лежал нормально вниз головой как в нашем случае); 2) с 2,5-3-х месяцев у нас хорошее такое слюноотделение, хотя сейчас поуменьшилось (что так же возникает из за раздражения ствола, хотя нам все говорили, что это нормально и часто бывает - зубы, не умеет глотать, так работают слюнные железы, скоро будут зубы и т.п.) и 3) заметила тонус пронаторов предплечий (больше на одной руке, вторая чуть чуть). Сказала, что можно попить Стугерон (снять спазмы задних мозговых артерий), но на последнем УЗИ первый врач "схалявила" (хотим его повторить) и не описала скорость кровотока в них на первом было 0,68, что не совсем нормально (норма 61). Но в целом сказала, что ребенок развит хорошо.
Хотим попросить местных врачей и специалистов оценить наше состояние по результатам в прикрепленных документах. Стоит ли нам волноваться? Какие действия еще предпринять, что бы не опоздать с лечением (если оно нужно). Судороги у нас вызывают сильную озабоченность (постоянно переживаем). Эпелиптолог сказала, что они могут спадать до 2-х лет - мы честно говоря не представляем как это будет влиять на качество жизни нашего малыша. Хотим их купировать (врач говорит, что принятие препаратов повлияет на развитие). Специалист по ДЦП сказала, что видела 2 раза такие случаи и что они прошли к году. Мы конечно будем следить за развитием малыша, но что можно еще предпринять кроме повторного ЭЭГ и посещения другого хорошего эпелиптолога? есть смысл пить Стугерон или он может навредить? Всем заранее спасибо за ответы.
Charlotte Dravet, Michelle Bureau
История и терминология
По нашим сведениям, на данный момент имеется 67 опубликованных в литературе случаев, из которых 10 описаны как рефлекторные (Dravet and Bureau 2002). Необходимо упомянуть о том, что в первом описании синдрома возраст дебюта приходился на период до 3 лет, в то время как последующие авторы описывали возможность и более позднего дебюта – вплоть до 4 лет 8 месяцев (Giovanardi Rossi et al 1997). Это означает, что один и тот же тип эпилепсии может дебютировать в разное время, но чаще в определенные периоды (Guerrini et al 1994).
Доброкачественная миоклоническая эпилепсия характеризуется появлением коротких миоклонических атак у здоровых младенцев в возрасте от ½ года до 3 лет. Более ранний дебют не типичен. Семейный анамнез по эпилепсии и фебрильным судорогам отягощен у 30% детей. Как правило, до появления миоклоний развитие ребенка протекает нормально, без признаков патологии. Однако имеются сообщения о наличии в анамнезе фебрильных судорог примерно у 20% случаев. Они обычно редкие, простые, как правило, предшествуют появлению миоклоний. У двоих пациентов были сопутствующие заболевания, у одного - синдром Дауна (Dravet and Bureau 2002), а у другого – диабет (Colamaria et al 1987).
В начале заболевания развитие остается нормальным, и эти движения не расцениваются родителями и педиатрами как патологические.
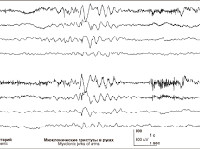
Интериктальная ЭЭГ может оставаться совершенно нормальной. Но миоклонии всегда сопровождаются на ЭЭГ быстрой генерализованной спайк-волновой или полиспайк-волновой активностью, частотой более 3 Гц, более или менее регулярной, длящейся от 1 до 3 секунд. Во время засыпания обычно наблюдается усиление миоклоний, которые обычно (но не всегда) прекращаются во сне. Ритмическая фотостимуляция также может провоцировать миоклонии.
Полиграфические записи демонстрируют связь между возникновением миоклоний и появлением на ЭЭГ разрядов спайк-волн или полиспайк-волн. Миоклонии короткие – от 1 до 3 секунд ) и обычно изолированные. За миоклониями может следовать короткий период атонии. Иногда после атаки может следовать произвольное движение, которое выглядит нормальным мышечным сокращением.
Интериктальная ЭЭГ детей соответствует возрасту. Спонтанные разряды спайк-волн регистрируются редко, может отмечаться медленноволновая активность над центральными областями. Если ритмическая фотостимуляция вызывает спайк-волны, последние всегда сопровождаются миоклониями. При записи сна регистрируется его нормальное разделение по фазам, во время REM-сна могут отмечаться генерализованные разряды спайк-волн.
У детей, страдающих доброкачественной миоклонической эпилепсией, другие типы приступов, в частности, абсансы или тонические приступы, не развиваются, даже если дети не получают лечение. Неврологический статус – без изменений. Интериктальный миоклонус описан только у 6 пациентов (Giovanardi Rossi et al 1997). При исследовании КТ или МРТ патологических изменений не было выявлено.
Прогноз зависит от своевременности диагностики и назначения лечения. Если не лечить, у пациента продолжаются миоклонические приступы, что может сказаться на психомоторном развитии и привести к появлению расстройств поведения. Миоклонии легко контролируются монотерапией вальпроатами, и ребенок может дальше развиваться в соответствии с возрастными нормами.
Ясного представления о механизмах развития синдрома на сегодняшний день нет. Генетика – не известна. Случаи редки сами по себе, семейных случаев не описано. Генетические связи с другими формами идиопатических генерализованных эпилепсий не установлены. Delgado-Escueta в своем исследовании не нашел ни одного случая юношеской миоклонической эпилепсии среди членов семей 24 пациентов, страдающих доброкачественной миоклонической эпилепсией младенчества (Delgado-Escueta et al 1990). Arzimanoglou описал случай заболевания второго мальчика в семье, старший брат которого страдал типичной формой миоклонических-астатических приступов, – синдромом Дузе. Этот случай поднимает вопрос о взаимосвязи двух синдромов в рамках одной большой группы идиопатических генерализованных эпилепсий детского возраста. Biondi описал случай с пациентом, у других членов семьи которого было подозрение на эпилепсию (Biondi et al 1991). У его отца и 2 сестер в ЭЭГ сна регистрировались короткие вспышки генерализованных спайк-волн.
Согласно данным немногочисленных эпидемиологических исследований, доброкачественная миоклоническая эпилепсия младенчества составляет меньше 1% всех эпилепсий (Loiseau et al 1991; Centre Saint-Paul 1997, неопубликованные данные), но около 2 % эпилепсий, начинающихся в первые 3 года жизни (Dalla Bernardina et al 1983), и 2% от всех идиопатических генерализованных эпилепсий (Centre Saint-Paul 1997, неопубликованные данные).
Нет данных о возможности профилактики синдрома.
Когда на первом году жизни у ребенка начинаются миоклонии, диагноз, который приходит на ум, это криптогенные инфантильные спазмы. Клинически спазмы отличаются от миоклоний. Они более интенсивные и вызывают отчетливое сгибание всего тела, что никогда не наблюдается при доброкачественной миоклонической эпилепсии. Помимо единичных, изолированных спазмов, у ребенка всегда наблюдаются также серии спазмов. На полиграфических записях инфантильных спазмов хорошо виден типичный паттерн короткого тонического сокращения, хорошо описанный Fusco и Vigevano, миоклонии редко носят продолжительный характер (Fusco and Vigevano 1993). Отличается и иктальная ЭЭГ – нет быстрой генерализованной спайк-волновой активности. Для нее характерны: внезапное уплощение ритма после гипсаритмии с наложением (или без) быстро-волновой активности, высокоамплитудные медленные волны, сменяемые уплощением, или даже отсутствие видимых изменений. Инфантильные спазмы всегда сопровождается изменением поведенческой активности, снижением контакта, нарушением психомоторного развития, вплоть остановки и даже регрессии. На интериктальных ЭЭГ всегда регистрируются патологические изменения – как истинная гипсаритмия, так и ее модифицированная форма, или фокальные нарушения; при этом никогда не регистрируются изолированные или короткие вспышки билатерально-синхронных спайк-волн, как при доброкачественной миоклонической эпилепсии.
В случаях, когда после нескольких обследований и психомоторное развитие, и ЭЭГ (как во время сна, так и бодрствования) остаются нормальными, даже если приступы напоминают инфантильные спазмы, то можно предположить у ребенка доброкачественный неэпилептический миоклонус (Lombroso and Fejerman 1977). У этих пациентов и в иктальной ЭЭГ нет изменений (Dravet et al 1986; Pachatz et al 1999).
На первом году жизни может дебютировать также тяжелая миоклоническая эпилепсия младенцев, но она всегда начинается с продолжительных, повторяющихся фебрильных судорог (но не с изолированных миоклоний), при этой форме всегда страдает психомоторное развитие (Dravet and Bureau 2002).
Наконец, необходимо исключать и другие эпилепсии, которые могут дебютировать в первые 3 года жизни и проявляющиеся главным образом миоклониями, имеют при этом различный прогноз. Они представляют собой различные сочетания других типов приступов, с фокальными измененими на ЭЭГ, задержкой психомоторного развития, плохим ответом на терапию АЭП и неоднозначным прогнозом (Dravet 1990).
Диагностический алгоритм довольно простой. Необходим качественный сбор анамнеза и повторные записи полиграфической видео-ЭЭГ для того, чтобы доказать наличие миоклонических атак с генерализованными разрядами спайк-волн. Миоклонии либо спонтанные, либо возникают в ответ на звук, прикосновение или ритмическую фотостимуляцию, а также при засыпании. Во время ЭЭГ сна можно увидеть некоторую активацию разрядов без изменения их морфологии, появление быстрых ритмов и фокальных нарушений. Нейровизуализация полезна (но необязательна), чтобы подтвердить отсутствие структурных нарушений головного мозга. Нейропсихологическое тестирование необходимо для проверки отсутствия нарушений психомоторного развития.
По определению прогноз благоприятный, миоклонические приступы прекращаются при условии назначения соответствующего лечения – монотерапии вальпроатами. В одном из исследований сообщалось, что только 5 пациентам было необходимо добавление второго препарата для достижения контроля над приступами (Giovanardi Rossi et al 1997). Имеющиеся данные по длительности наблюдения различны – от 9 месяцев до 27 лет. У 10 пациентов появились редкие генерализованные тонико-клонические судороги, не сочетаясь с миоклониями, из них у 3 они возникли после отмены препарата, у остальных – в подростковом возрасте (Dravet and Bureau 2002). Приступы, провоцируемые звуком или прикосновением, легче поддаются контролю, чем спонтанные. И наоборот, фотосенситивность труднее контролируется, и может регистрироваться на протяжении нескольких лет после прекращения приступов.
Прогнозировать исход по психическому развитию сложнее. В большинстве случаев прогноз довольно благоприятный. Однако в рамках продолжительных исследований описаны 12 пациентов, у которых отмечались умеренная задержка психического развития, личностные расстройства или легкие нарушения поведения (Colamaria et al 1987; Todt and Muller 1992; Giovanardi Rossi et al 1997; Dravet and Bureau 2002). Ни один из этих пациентов не был госпитализирован для специализированного лечения. Благоприятный прогноз по психологическим и когнитивным функциям также зависит от своевременности диагноза, назначения адекватного лечения и помощи со стороны родственников. Но существуют и противоположные факторы, как то - проблемы в семье и неблагоприятные особенности взаимоотношения между матерью и ребенком.
В первую очередь назначается монотерапия вальпроатами, лучше в инъекциях, т.к. дети могут отказываться пить сироп. Необходимо тщательно контролировать его уровень в плазме крови, поскольку нерегулярность приема может привести к возобновлению приступов и имитировать резистентную форму. Суточная дозировка 30 мг/кг, как правило бывает достаточной, но иногда необходимо и увеличивать дозу (Lin et al 1998).. Вальпроат также эффективен против фебрильных судорог. Если миоклонии не уходят полностью на вальпроате, можно попробовать добавить бензодиазепин (клобазам или нитразепам) или этосуксимид, либо пересмотреть диагноз. Терапию, если она переносится хорошо, необходимо продолжать в течение 3-4 лет с начала приступов, более продолжительно в случаях с фотосенситивностью. Если приступы носят исключительно рефлекторный характер, можно обойтись без приема вальпроата либо прекратить терапию раньше. В случае возникновения одного генерализованного тонико-клонического приступа в подростковом возрасте, возможно, потребуется еще один, короткий курс лечения.
Список литературы
Arzimanoglou A, Prudent M, Salefranque F. Epilepsie myoclono-astatique et epilepsie myoclonique benigne du nourrisson dans une meme famille: quelques reflexions sur la classification des epilepsies. Epilepsies 1996;8:307-15.**
Beaumanoir A, Blume W. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet Ch, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd, 2002:113-35.
Biondi R, Sofia V, Tarascone M, Leocata R. Epilessia mioclonica benigna dell’infanzia: contibuto clinico. Boll Lega Ut Epil 1991;74:93-4.
Colamaria V, Andrighetto G, Pinelli L, Olivieri A, Alfieri P, Dalla Bernardina B. Iperinsulinismo, ipoglicemia ed epilessia mioclonica benigna del lattante. Boll Lega It Epi 1987;58/59:231-3.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Dalla Bernardina B, Colamaria V, Capovilla G, Bondavalli S. Nosological classification of epilepsies in the first three years of life. In: Nistico G, Di Perri R, Meinardi H, editors. Epilepsy: an update on research and therapy. New-York: Alan Liss, 1983:165-83.
Delgado-Escueta AV, Greenberg D, Weissbecker K, et al. Gene mapping in the idiopathic generalized epilepsies: juvenile myoclonic epilepsy, childhood absence epilepsy, epilepsy with grand mal seizures, and early childhood myoclonic epilepsy. Epilepsia 1990;31(suppl 3):S19-29.
Doose H. Myoclonic astatic epilepsy of early childhood. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence, 2nd ed. London: John Libbey Eurotext Ltd., 1992:103-14.
Dravet C. Les epilepsies myocloniques benignes du nourrisson. Epilepsies 1990;2:95-101.
Dravet C, Bureau M. L’epilepsie myoclonique benigne du nourrisson. Rev Electroenceph Neurophysiol Clin 1981;11:438-44.
Dravet C, Bureau M, Giraud N, Roger J, Gobbi G, Dalla Bernardina B. Benign myoclonus of early infancy or benign non-epileptic spasms. Neuropediatrics 1986;17:33-8.
Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. In: Roger J, Bureau M, Dravet Ch, Genton P, CA Tassinari, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd., 2002:69-79.**
Fusco L, Vigevano F. Ictal clinical and electroencephalographic findings of spasms in West syndrome. Epilepsia 1993;34:671-8.
Giovanardi Rossi P, Parmeggiani A, Posar A, Santi A, Santucci M. Benign myoclonic epilepsy: long-term follow-up of 11 new cases. Brain Dev 1997;19:473-9 **
Guerrini R, Dravet CH, Gobbi G, Ricci S, Dulac O. Idiopathic generalized epilepsies with myoclonus in infancy and childhood. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalized epilepsies: clinical, experimental, and genetic aspects. London: John Libbey Eurotext Ltd., 1994:267-80. **
Lin YP, Itomi K, Takada H, et al. Benign myoclonic epilepsy in infants: video-EEG features and long-term follow-up. Neuropediatrics 1998;29:268-71.
Loiseau P, Duche B, Loiseau J. Classification of epilepsies and epileptic syndromes in two different samples of patients. Epilepsia 1991;32:303-9.
Lombroso CT, Fejerman N. Benign myoclonus of early infancy. Ann Neurol 1977;1:138-43.
Pachatz C, Fusco L, Vigevano F. Benign myoclonus of early infancy. Epil Disord 1999;1:57-61.
Ricci S, Cusmai R, Fusco L, Cilio R, Vigevano F. Reflex myoclonic epilepsy of the first year of life. Epilepsia 1995;35:47.**
Todt H, Muller D. The therapy of benign myoclonic epilepsy in infants. In: Degen R, Dreifuss FE, editors. Epilepsy research. Suppl 6. The benign localized and generalized epilepsies in early childhood. Amsterdam: Elsevier, 1992:137-9.
Vigevano F, Cusmai R, Ricci S, Watanabe K. Benign epilepsies of infancy. In: Engel Jr J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers, 1997:2267-76.
Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – зависящая от возраста форма идиопатической эпилепсии, которая характеризуется генерализованными миоклоническими припадками. Этиология детально не изучена. Патология проявляется мышечными сокращениями верхних конечностей, шеи и головы длительностью в 1-3 сек. с частотою 2-3 раза в день. Общее состояние ребенка и его психофизическое развитие нарушается редко. Диагностика направлена на определение спайк- или полиспайк-волн на ЭЭГ. Основное лечение – медикаментозная монотерапия. Препараты выбора – вальпроаты, при их неэффективности используются бензодиазепины или производные сукцинимида.
- Причины ДМЭМ
- Симптомы ДМЭМ
- Диагностика ДМЭМ
- Лечение ДМЭМ
- Прогноз и профилактика ДМЭМ
- Цены на лечение
Общие сведения
Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – редкая форма эпилепсии в педиатрии. Характерна только для определенной возрастной категории. Впервые заболевание выделено как отдельная нозологическая форма в 1981 году Дарве и Биором. Патология составляет менее 1% от всех форм эпилепсий и порядка 2% от ее идиопатических генерализованных форм. На данный момент в литературе описано около 100-130 случаев данного заболевания. ДМЭМ наблюдается у детей от 6 месяцев до 3 лет, в редких случаях возникает в возрасте до 5 лет. Представители мужского пола болеют в 1,5-2 раза чаще. Патология, как правило, хорошо поддается лечению и полностью купируется в старшем возрасте (в основном – после 6 лет). Осложнения в виде отставания в психомоторном развитии возникают редко и только при отсутствии терапии.
Причины ДМЭМ
Доброкачественная миоклоническая эпилепсия младенчества относится к числу генетически детерминированных заболеваний, передающихся по полигенному типу наследования. Является малоисследованной патологией, поскольку встречается довольно редко. ДМЭМ входит в группу идиопатических генерализованных эпилепсий, однако связи с другими нозологиями из этой группы не установлено. На данный момент неизвестно, мутация каких генов приводит к развитию ДМЭМ.
При сборе семейного анамнеза выясняется, что родители 40% больных страдают или страдали эпилепсией либо фебрильными припадками. Патогенетически развитие миоклонических атак обусловлено возникновением разряда быстрых генерализованных спайк-волн (СВ) или полиспайк-волн (ПСВ). Их частота составляет 3 Гц или более, а длительность – 1-3 сек. Волны формируются в лобных или теменных участках коры головного мозга. Сами атаки могут быть спонтанными или возникающими на фоне определенных (звуковых, тактильных или ритмичных световых) раздражителей.
Симптомы ДМЭМ
ДМЭМ диагностируется в возрасте от 6 месяцев до 3 лет. Развитие ребенка до появления первых миоклонических припадков проходит нормально. Примерно у 20% детей проявляются редкие судороги при рождении или в неонатальном периоде. Общее состояние пациента страдает редко, нарушения неврологического статуса не выявляются. Первые миоклонические атаки поражают верхние конечности, шею и голову, редко – ноги. Они могут иметь разную интенсивность, в том числе – у одного и того же ребенка во время разных эпизодов. Выраженность колеблется от еле заметных подергиваний до видимых фибрилляций.
Частота припадков составляет 2-3 раза в сутки с разными временными интервалами. Длинных серий атак не наблюдается. Возможна провокация атаки громким звуком, тактильной или ритмичной световой стимуляцией. После каждого эпизода может наблюдаться рефрактерный период длительностью от 20 до 120 сек. В этом временном промежутке даже интенсивная стимуляция не вызывает новый приступ. При этом часто наблюдается мышечная атония. Для заболевания характерно усиление миоклонических приступов при засыпании (дремоте) и их исчезновение в фазе медленного сна.
При тяжелых формах возможна генерализация судорог, сопровождающаяся потерей равновесия, внезапным выпадением предметов из рук, редко – расстройствами сознания. В процесс иногда вовлекаются межреберные мышцы, передняя брюшная стенка и диафрагма, из-за чего нарушается дыхание и может выслушиваться экспираторный шум. Для ДМЭМ характерно увеличение интенсивности клинических проявлений до определенного возраста и их последующее полное исчезновение. При длительном течении заболевания возможно отставание в психомоторном развитии. Трансформация в другие формы приступов, в том числе в абсансы, не происходит даже на фоне отсутствия специфического лечения.
Диагностика ДМЭМ
Диагностика доброкачественной миоклонической эпилепсии младенчества заключается в сборе анамнестических данных и проведении инструментальных методов исследования. Физикальное обследование ребенка в интериктальный период неинформативно. Лабораторные тесты каких-либо отклонений от возрастной нормы не выявляют. Наибольшую диагностическую ценность имеет повторная полиграфическая видеоэлектроэнцефалография (видео-ЭЭГ), которая позволяет обнаружить спайк-волны и доказать наличие миоклонических приступов. При необходимости проводится провокационная проба с ритмической световой или тактильной стимуляцией.
Вне приступов (а изредка – во время них) данные ЭЭГ остаются в пределах нормы, спонтанные спайк-волны возникают редко. Во время медленного сна возможно усиление разрядов в коре головного мозга при сохранении их нормальной структуры, возникновение быстрых ритмов или формальных изменений. В быструю фазу (REM-сон) могут фиксироваться генерализованные разряды спайк-волн. С целью исключения органической патологии могут назначаться нейросонография, компьютерная и магнитно-резонансная томография. При наличии клинической симптоматики на протяжении длительного периода проводится оценка психомоторного развития.
Дифференциальная диагностика ДМЭМ осуществляется с криптогенными детскими судорогами, доброкачественным неэпилептическим миоклонусом, синдромом Леннокса-Гасто и миоклонически-астатической эпилепсией раннего детского возраста.
Лечение ДМЭМ
Лечение ДМЭМ, как правило, проводится в амбулаторных условиях, исключением являются частые и тяжелые миоклонические атаки, требующие постоянного наблюдения. Показана медикаментозная терапия при помощи противоэпилептических средств. Первую линию составляют препараты из группы вальпроатов (натрия вальпроат). Важную роль играет подержание стабильной концентрации действующего вещества в крови. Нерегулярное введение назначенных средств провоцирует новые приступы и формирование резистентности к дальнейшей терапии данным медикаментом. При недостаточной эффективности вальпроатов показаны препараты из группы бензодиазепинов (нитразепам) или производных сукцинимида (этосуксимид). Терапевтический курс предполагает лечение на протяжении 3-4 лет с момента возникновения первых припадков.
При выраженной чувствительности к ритмичным световым раздражителям длительность курса увеличивается. При минимальной активности приступов или их исключительно рефлекторном характере лечение может проводиться в сокращенные сроки или не назначаться вовсе. При рецидиве миоклонических атак в старшем возрасте после пройденного лечения рекомендован упрощенный вариант аналогичного терапевтического курса. Обязательным моментом является психологическая поддержка семьи, непосредственно влияющая на эффективность лечения и направленная на исключение триггеров для ребенка.
Специфической профилактики для доброкачественной миоклонической эпилепсии младенчества не разработано. Прогноз в большинстве случаев благоприятный, заболевание, как правило, заканчивается полным выздоровлением ребенка. Миоклонические атаки, возникающие на фоне звуковой или тактильной стимуляции, прогностически более благоприятны, чем спонтанные. Переход в другие формы эпилепсии нехарактерен. Острый период, при котором наблюдаются выраженные припадки, в среднем длится менее 12 месяцев. Более чем у 53% детей в возрасте 6 лет все симптомы ДМЭМ полностью исчезают. Примерно у 14% в дальнейшем наблюдаются задержка психического развития или нарушения в поведении, из-за чего пациенты вынуждены получать образование в специализированных учебных заведениях. Частота осложнений напрямую зависит от своевременности диагностики, эффективности проводимого лечения и психологического климата в семье, в первую очередь – взаимоотношений между ребенком и матерью.
Читайте также: