
Все мозжечковые атаксии луи бар

Атаксией называется нарушение согласованности движений в разных группах мышц, не сопровождаемое развитием мышечной слабости. При этом движения сохраняются, но происходит их раскоординация. В результате этой патологии могут нарушиться многие функции организма, в частности движения глаз, речь, ходьба, мелкая моторика, глотание.
Классификация
Атаксия бывает наследственной и приобретённой. Также заболевание классифицируют по характеру нарушений и клиническим признакам.
- Статическая атаксия проявляется в виде нарушения равновесия в положении стоя.
- Динамическая характеризуется как дискоординация движений.
- Статодинамическая сочетает признаки обоих видов.

Также выделяют особый вид, не связанный с органическими патологиями, – интрапсихическая или психогенная атаксия. Этот вид характеризуется расщеплением психических функций и их речевого или мимического выражения.
Встречается при шизофрении. Выражается в вычурных движениях при ходьбе и стоянии. Больной ходит, не разгибая ног, перекрещивая их.
Заболевания, которые могут привести к атаксии:

- Рассеянный склероз;
- Сосудистые заболевания головного мозга (ишемический и геморрагический инсульты);
- Опухолевые заболевания коры и подкорковых структур;
- Травмы черепа и вещества головного мозга;
- Врождённые пороки развития черепной коробки и мозга;
- Демиелинизирующие заболевания нервов;
- Заболевания уха или преддверно-улиткового нерва;
- Отравления некоторыми лекарственными препаратами;
- Дефицит витамина В12;
- Гипотиреоз;
- Хронический алкоголизм;
- Наследственные патологии.
Наследственные атаксии
Это прогрессирующее наследственное заболевание. Для данной патологии характерна ранняя манифестация. Болеют чаще мужчины, наследуется по аутосомно-доминантному типу. Основным проявлением является сочетание мозжечковой и сенситивной атаксии. Для семейной атаксии характерно ослабление рефлексов и снижение тонуса мышц, появление нистагма. Также возможно врождённое слабоумие вплоть до дебильности. Помимо неврологических изменений, атаксия Фридрейха характеризуется поражением сердца (миокардиодистрофией), снижением слуха, деформацией позвоночника. К симптомам присоединяются одышка, тахикардия, приступообразные боли в сердце.
Ниже следует видеолекция об этом заболевании:
Представляет собой прогрессирующее заболевание, передающееся по аутосомно-доминантному типу, и сочетает в себе все признаки мозжечковой атаксии. Проявляется гипоплазией мозжечка и атрофией варолиева моста и олив. Заболевание манифестирует после 30 лет, раньше – крайне редко. Иногда клиническая картина схожа с семейной атаксией Фридрейха. Клиническая картина: усиление рефлексов, повышение тонуса мышц, уменьшение силы в конечностях. Изредка могут фиксироваться патологические изменения со стороны органа зрения (снижение остроты, сужение полей, атрофия зрительного нерва).
Быстро прогрессирующее аутосомно-рецессивное заболевание, проявляющееся недоразвитием тимуса (вилочковой железы) и дисгаммаглобулинемией. Первые признаки проявляются еще в детском возрасте. Клиническая картина сходна с мозжечковой атаксией. Больные склонны к частым и рецидивирующим инфекционным заболеваниям, телеангиэктазиям (пятнам на коже). Уровень интеллекта снижен. Рефлексы замедленные, возможно появление гипер- или гипокинезов. Вследствие дефицита гуморального звена иммунитета повышен риск появления новообразований.
Причины и признаки болезни Вильсона-Коновалова описаны здесь.
Диагностика всех видов атаксий основывается на данных анамнеза, общего осмотра, данных лабораторно-инструментальных исследований.
При сборе анамнеза отмечают время появления первых симптомов, наследственную предрасположенность, перенесённые заболевания в течение жизни. Во время общего осмотра оценивают рефлексы, тонус мышц, зрение, слух, выполняют координационные пробы.

- Общий и биохимический анализы крови и мочи с целью выявления патологий в обмене веществ и нарушении гомеостаза;
- Спиномозговая пункция с последующим анализом ликвора;
- Проведение ряда генетических тестов для определения мутаций;
- Электронейромиография, которая позволяет выявить нарушения в проведении нервно-мышечного импульса;
- Магнитно-резонансная томография головного или спинного мозга;
- Электроэнцефалография позволит выявить патологическую активность участков мозга;
- ДНК-диагностика при наследственных формах;
- Биохимический скрининг для выявления заболеваний, провоцирующих атаксию.
Лечение
Лечение атаксий направлено на общее укрепление организма, повышение иммунитета и максимально долгое сохранение двигательной активности пациентов. Также возможно практически полностью вылечить пациента, при условии устранения причины атаксии (опухоли, абсцесса, снижения давления, детоксикации).
В общеукрепляющей терапии относят применение нейровитаминов, витамина Е, янтарной кислоты, а также препаратов, направленных на коррекцию иммунитета, нормализацию артериального давления, улучшение микроциркуляции мозга.
При инфекционных поражениях применяется антибиотикотерапия.
При демиелинизирующих заболеваниях показаны гормональные препараты и плазмаферез.

При отсутствии лечения прогноз как для трудоспособности, так и для жизни неблагоприятный. Происходит постепенное разрушение нервно-психической системы, что влияет как на общее самочувствие больного, так и на возможность социальной активности. Усугубляют ситуацию инфекционные заболевания, отравления, травмы.
Улучшить прогноз заболевания может его раннее выявление и назначение адекватной терапии, а также помощь больным в адаптации. Это позволяет продлить период активной трудоспособной жизни.
Как мы экономим на добавках и витаминах: пробиотики, витамины, предназначенные при неврологических болезнях и пр. и мы заказываем на iHerb (по ссылке скидка 5$). Доставка в Москву всего 1-2 недели. Многое дешевле в несколько раз, нежели брать в российском магазине, а некоторые товары в принципе не найти в России.

- 7 Июля, 2018
- Неврология
- Гладких Ася
Синдром Луи-Бар относится к редким иммунодефицитным нейродегенеративным генетическим заболеваниям, проявляющимся в виде мозжечковой атаксии, которая вызывает паралич тяжелой формы. Для атаксии характерно нарушение координации движений, а для телеангиэктазии — расширение кровеносных сосудов. Два этих признака – это отличительные черты синдрома. Поэтому у заболевания имеется второе название — атаксия-телеангиэктазия.
Тип наследования
Передается данный недуг по аутосомно-рецессивному типу. Риск заболевания ребенка, рожденного в семье, в которой один больной родитель, составляет 50/50. Согласно статистическим данным, заболевание мало распространено, из сорока тысяч оно приходится на одного человека.

Данная патология характеризуется врожденным неправильным иммунным состоянием человеческого организма. Происходит поражение Т-звена в генетической цепочке. Затем болезнь проявляется в виде аномальных форм по всему организму. В связи с поражением иммунитета люди, которые больны синдромом Луи-Бар, подвержены частым инфекционным заболеваниям. Кроме этого, могут возникнуть злокачественные онкологические опухоли по всему телу.
В случаях, когда синдром проявляется у новорожденного ребенка, шансы на выживание практически отсутствуют. Причем во многих случаях не удается даже своевременно и правильно поставить диагноз.
Причины патологии (мнение специалистов)
Синдром Луи-Бар в различных классификациях рассматривают как спинно-мозжечковую дегенерацию или как факоматоз (данный термин предложили в качестве обозначения заболеваний с совмещенным поражением нервной системы и покровов кожи — врожденных нейро-эктомезодермальных дисплазий). Причиной является мутация АТМ гена, активирующего аутоиммунные процессы, которые приводят к уничтожению клеток всего организма, в том числе и головного мозга. Генетические отклонения проявляются уже во внутриутробном периоде.
Данная болезнь в равной степени выявляется и у мужчин, и у женщин. Она стремительно развивается, поражая в первую очередь ЦНС и покровы кожи. Недуг имеет способность полностью изменить или разрушить ткани мозжечка, но поражает его ядро.
Синдром Луи-Бар – это иммунодефицитное состояние, основанное на гипоплазии тимуса и дефиците IgA и IgE. Это означает, что нарушается иммунная деятельность клеточного и гуморального звеньев. Это может провоцировать часто повторяющиеся заболевания дыхательных путей, носящие инфекционный характер, ЖКТ и кожных покровов. Типичная гипоплазия вилочковой железы может быть дополнена гипо/атрофией лимфатических узлов и в целом лимфатического аппарата, а также селезенки и пищеварительного канала.
Ослабленному иммунитету не по силам выстоять даже перед несерьезной инфекцией, кроме того, он становится чувствительным перед злокачественными новообразованиями в лимфатической системе.
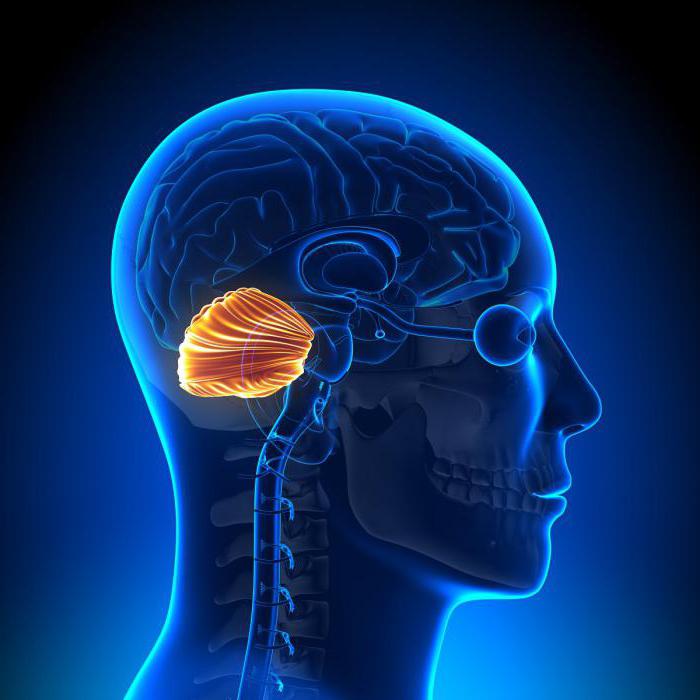
Клиника синдрома
Это врожденное заболевание встречается довольно редко. Первые симптомы могут проявиться в возрасте 3-х месяцев и далее - до 3-х лет. По мере роста больного проявления становятся более выраженными.
Телеангиэктазия проявляется, как правило, после признаков атаксии, при достижении больным возраста 4-6 лет. Иногда случается так, что симптомы заметны уже в первые недели жизни. Телеангиэктазии характеризуются проявлениями изначально на глазных яблоках по типу бульбарной коньюктивы, после они начинают распространяться на веки и лицо.
Характерная симптоматика синдрома Луи-Бар:
- нарушается координация движений (после 3-лет);
- страдает психика и замедляется или полностью останавливается развитие;
- изменяется цвет кожи под ультрафиолетовыми лучами;
- образуются белые пятна на теле;
- расширяются кровеносные сосуды в области внутренней стороны коленей и локтей;
- появляется ранняя седина;
- наблюдается повышение чувствительности к рентгеновским лучам;
- прогрессируют тяжелые инфекции дыхательных путей, ушей (у 80% больных);
- отсутствуют рефлексы в мышцах глаз;
- неправильно развивается вилочковая железа, иногда она вовсе отсутствует;
- наличие лимфоцитопении у 1/3 больных;
- половое развитие задерживается, наступает ранняя менопауза.
Это характерно для неврологических болезней.

Проявления симптомов на кожных покровах наблюдаются при данном заболевании в 100% случаев. Возникновение признаков другого типа – сухость кожи, кератоз кожных покровов конечностей, пигментация на лице – можно встретить лишь в половине случаев. Кожные проявления не являются специфичным симптомом для патологии, но это первый наглядный признак болезни, который очень важен при своевременном и правильном диагностировании и лечении. Зачастую именно благодаря ему можно своевременно и правильно поставить диагноз.
Диагностирование синдрома
По мнению специалистов, синдром Луи-Бар распознать может быть сложно по причине сочетания в нем признаков других генетических заболеваний, которые скрывают настоящие симптомы. В большинстве случаев выявить и провести диагностику недуга возможно только по истечении продолжительного лечения инфекционных заболеваний, не дающего результатов.
Чтобы не ошибиться и установить правильный диагноз, больной должен проконсультироваться не у одного медицинского специалиста. Это может быть иммунолог, дерматолог, офтальмолог, онколог, отоларинголог. Нужно проанализировать пройденные процедуры, изучить анализы, прочитать заключения специалистов после консультаций. И только после этого окончательный диагноз сможет поставить врач-невролог. Кроме того, он должен назначить лабораторные исследования, другие процедуры и тесты, это поможет не ошибиться в формулировках.

Что выявляют на осмотре?
При осмотре врач должен обратить особое снимание на:
- замедление темпов развития;
- пигментацию кожных покровов;
- нарушение или отсутствие сухожильных рефлексов;
- нарушение роста;
- уменьшенные размеры миндалин, лимфатических узлов.
На что направлена диагностика?
Список лабораторных анализов, которые могут быть назначены, нацелены на:
- установление уровня белка α-фетопротеина (при заболевании его содержание повышено);
- снижение уровня лейкоцитов;
- исследование уровня иммуноглобулина в крови (при болезни уровни иммуноглобулина А и Е сильно снижены);
- выявление генетических мутаций;
![]()
- определение переносимости глюкозы;
- ультразвуковое обследование вилочковой железы.

Кроме этого, проводится МРТ головного мозга и мозговых структур (при синдроме заметно увеличен четвертый желудочек и наблюдаются изменения патологического уровня в мозжечке — дегенерация мозжечковых клеток). Рентгенография грудной клетки поможет исключить пневмонию.
Терапия заболевания
На сегодняшний день медицина не в состоянии бороться с таким тяжелым генетическим врожденным заболеванием, как синдром Луи-Бар. Решение данного вопроса является одной из главных задач экспериментальной медицины в области генетики. Как правило, лечение заключается в замедлении прогрессирования заболевания и уменьшению его симптомов.
Терапия может быть назначена только врачом-неврологом, причем каждый отдельный случай рассматривается индивидуально для каждого пациента. При этом учитываются этиология, патогенез, стадия заболевания. Чтобы продлить жизнь пациента, специалист назначает особую иммунотерапию с разными дозами Т-активина и гамма-глобулина. Вместе с тем необходимо обязательно принимать витамины, чтобы поддерживать правильное функционирование организма.
Курс антибиотиков
Пациент проходит назначенный ему курс терапии антибиотиками, чтобы побороть вторичную инфекцию, носящую бактериальный характер. Также больному прописываются физиотерапевтические процедуры.
Химиотерапия
Если врач при диагностировании обнаруживает злокачественные новообразования, он назначает химиотерапию, лучевую терапию или ставит вопрос о хирургическом вмешательстве. Если больной страдает сахарным диабетом, то в таком случае ему назначают инсулин и противодиабетические препараты. К неврологическим болезням нужно подходить со всей серьезностью.
Как спрогнозировать синдром?
Так как заболевание является генетическим, разрушает частично или полностью на клеточном уровне иммунитет, носит патологический характер и не излечивается, то естественная полноценная жизнедеятельность становится практически невозможной.

Прогнозы при этом заболевании врожденного характера неутешительны. Многие больные умирают спустя 5-8 лет после появления первых симптомов от патологий дыхательной системы, которые носят инфекционный характер (например, от воспаления легких) или от образовавшихся в организме злокачественных опухолей. Как правило, больные данным синдромом живут в среднем до 14-15 лет, но бывают редкие исключения, когда при благоприятных условиях жизни пациенты могут дожить до 40 лет.
Профилактических или предупредительных мер заболевания нет по той простой причине, что невозможно оказать влияние на генетику эмбриона в утробе матери.
В статье были рассмотрены характерные проявления синдрома Луи-Бар.
Синонимы синдрома Луи-Бар. S. Boder—Sedgwick. Це-фало-глазо-кожная телеангиэктазия. Мозжечково-глазо-кожная телеангиэктазия. Телеангиэктатическая атаксия. Атрофия мозжечка с глазо-кожными телеангиэктазиями и бронхоэктазиями. Синдром телеангиэктазии и атаксии.
Определение синдрома Луи-Бар. Редкий факоматоз у детей. Относится к нейро-кожным синдромам.
Авторы. Louis-Bar Denise — современный французский врач. Впервые описала синдром в 1941 г. В 1957—1958 гг. Boder и Sedgwick опубликовали 8 новых наблюдений и впервые привели данные аутопсии.
Симптоматология синдрома Луи-Бар:
1. Впервые проявляющиеся в раннем детском возрасте и медленно прогрессирующие мозжечковые атаксия, абазия и астазия; ко времени полового созревания свободные походка и стояние, как правило, невозможны. Одновременно развиваются расстройства речи (монотонная скандированная речь или регулярная дизартрия), также носящие прогрессирующий характер.
2. Отсутствие пирамидных знаков, рефлексы нормальные или ослаблены. Мышечный тонус (после первичного ригидноподобного повышения) обычно снижен. Нормальная чувствительность. Отсутствие парезов.
3. Медленно развивающиеся симметричные телеангиэктазии кожи и слизистых оболочек, особенно кожи лица и конъюнктивы (ранний симптом, который может проявиться быстропроходящим конъюнктивитом!). Частое развитие бляшек цвета кофе с молоком, атрофии кожи лица, преждевременное поседение волос (в школьном возрасте).
4. Рецидивирующие легочные инфекции, иногда с развитием бронхоэктазий.
5. Гиперсаливация.
6. Малый рост и общая дистрофия.
7. К началу заболевания интеллектуальное развитие нормальное, позднее происходит задержка психического развития.
8. Пневмоэнцефалографические данные: признаки мозжечковой атрофии.
9. Атаксия — телеангиэктазия очень часто комбинируется с гипоплазией вилочковой железы, специфической дисгаммаглобулинемией (недостаточность гамма Ау, глобулина) и склонностью к злокачественным процессам в ретикулоэндотелиальной системе (лимфосаркома, ретикулезы и т. д.).
10. Прогноз плохой. Большинство наблюдавшихся до сих пор больных погибали в период полового созревания.
Этиология и патогенез синдрома Луи-Бар. Рецессивно-наследственное расстройство с генетически обусловленным торможением васкуляризации головного мозга. В одном случае была установлена транслокация между двумя акроцентрическими хромосомами группы 13—14—15 (Bijl, Jansen, Ossentjuk, 1963). До сих пор не ясно значение обнаруживаемого в отдельных случаях избыточного выделения с мочой полипептидов.
Патологическая анатомия. Первичнохроническая прогрессирующая мозжечковая дегенерация с патологическими изменениями клеток Пуркине и сморщиванием белого вещества, а также изменения вен (расширение, засстой, истончение стенок), особенно в области мягкой мозговой оболочки мозжечка, а также и больших полушарий.
Дифференциальный диагноз. В начальных стадиях: мозжечковая форма синдрома церебрального детского паралича. S. Friedreich I (см.). Опухоли мозжечка. S. Sturge—Weber (см.). S. v. Hippel—Lindau (см.). S. Werner (см.). S. Osier I (см.).
Синдром Луи-Бар (атаксия-телеангиэктазия) — наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
- Причины и патогенез синдрома Луи-Бар
- Клинические проявления синдрома Луи-Бар
- Диагностика синдрома Луи-Бар
- Лечение и прогноз синдрома Луи-Бар
- Цены на лечение
Общие сведения
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
Причины и патогенез синдрома Луи-Бар
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Клинические проявления синдрома Луи-Бар
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне или проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома. Особенностью онкопатологии в случае синдрома Луи-Бар является повышенная чувствительность пациентов к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при их лечении.
Диагностика синдрома Луи-Бар
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться: УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Лечение и прогноз синдрома Луи-Бар
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
В связи с отсутствием эффективных способов лечения синдром Луи-Бар имеет неблагоприятный прогноз как для выздоровления, так и для жизни. Больные этим заболеванием редко доживают до 20 лет. В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
Общая информация
Ввиду того, что в теле предусмотрено несколько отделов нервной системы, обеспечивающих равновесие и координацию, при возникновении атаксий врач, прежде всего, проводит обследование на предмет исключения сбоев в работе одного из них. То есть, в работе мозжечка, коры лобной доли, вестибулярного аппарата, проводников глубокой суставно-мышечной чувствительности, затылочной и височной доли головного мозга.
Обратите внимание! Атаксии – это патологии, которые влекут за собой раскоординацию движений. Иными словами, у человека с таким диагнозом нарушаются: речь, ходьба, мелкая моторика, глотание, движения глаз. Ее двигательная активность может становиться неплавной, прерывистой или вовсе усложняться.
Прогноз
Прогноз зависит от множества факторов:
- Причины атаксии;
- Возраста пациента;
- Наличия сопутствующих патологий;
- Формы и распространенности процесса.
Что именно ждет больного сказать трудно. Каждый случай рассматривают индивидуально. Если запускающий фактор крылся в опухоли мозжечка, ее удаляют хирургическим путем. После этого больного может ждать полное восстановление. Помощь в адаптации к новым условиям жизни заметно улучшает состояние пациентов, а также позволяет продлить их социализацию.
Отсутствие лечения приводит к потере трудоспособности. Прогноз жизни у таких больных неблагоприятный. Нарушения в нервной системе истощают пациента. Люди теряют нить к социализации и уходят в себя. А наслаивающиеся осложнения в виде инфекционных процессов, только ухудшают ситуацию. Ввиду этого при первых тревожных симптомах необходимо обращаться к доктору. Выполнение его назначений – ключ к более нормальной жизни для больных.
Автор статьи: Врач невролог Махеев Константин Олегович.
В первую очередь, диагностируется наследственная и приобретенная атаксия. Существует еще одна классификация – в зависимости от характера поражений. Согласно ей, патология может быть:
- статической, когда нарушается равновесие (человеку сложно спокойно стоять);
- динамической, когда отмечается нарушение движений, например, при ходьбе;
- статодинамической, когда проявляются признаки обоих видов.
В зависимости от места локализации поражения выделяют атаксию:
Обратите внимание! Медики выделяют также психогенную и интрапсихическую атаксию. В случае их развития у человека отмечается расщепление функций психики. Узнать этот вид заболевания можно по вычурным движениям при ходьбе: пациент идет, не разгибая и не перекрещивая ног.
Атаксии, которые носят наследственный характер, также подразделяются на несколько видов, а именно:
- Атаксия Пьера Мари – передается от родителей и характеризуется появлением симптомов мозжечковой атаксии, гипоплазии мозжечка, атрофии моста мозга. В группе риска – люди в возрасте от 30 – 35 лет. Патология подозревается при нарушениях мимики, речи, появлении трудностей в ходьбе, утрате способности быстро двигать руками, непроизвольных сокращениях мышц пальцев и конечностей, птозе – опущении верхнего века, появлении депрессии, страха.
- Атаксия Фридрейха. Патология, возникающая в кровном браке. Проявляется преимущественно нарушениями походки, когда человек при ходьбе расставляет ноги слишком широко. При прогрессировании болезни отмечается нарушение координации рук, мимики, когда происходит непроизвольное сокращение мышц лица, замедление речи, ухудшение слуха, снижение рефлексов. Если пациенту своевременно не оказать помощь, у него развиваются сердечно-ссудистые болезни, возрастает риск переломов и вывихов суставов. Часто патология сопровождается гормональными расстройствами, сахарным диабетом, половыми дисфункциями.
- Синдром Луи-Бар. Наследственная патология, признаки которой проявляются еще в раннем детстве, когда достигнув 9-летнего возраста, ребенок теряет способность ходить. Наряду с этим у него отмечается умственная отсталость, склонность к острым инфекционным заболеваниям – отитам, бронхитам, пневмониям, гипоплазия вилочковой железы. Часто на этом фоне возникает злокачественная опухоль, что усугубляет течение заболевания, делая его прогноз неблагоприятным.
Проявления: какие еще возможны?
Если развивается вестибулярная форма, ее можно заметить по частым позывам к тошноте и рвоте. У больного кружится голова, это ощущение становится сильнее, если повернуть голову, даже при плавном движении.
Корковая атаксия выражает себя неустойчивостью при движении, наиболее выраженной при поворотах, нарушениями восприятия запахов, а также психическими отклонениями. У больного пропадает хватательный рефлекс.
Причины атаксии
Причинами возникновения патологии являются:
Обратите внимание! Острые формы атаксии возникают, как правило, на фоне системной красной волчанки, эпилепсии, серповидно-клеточной анемии.
Типы и формы: мозжечковая
Наследственная болезнь может быть врожденный и не склонной к прогрессу, аутосомно-рецессивной и рецессивной, при которой недостаточность мозжечка постепенно прогрессирует. Выделяют форму Баттена, врожденную форму, при которой развитие ребенка замедляется, но в будущем пациенту удается адаптироваться. Поздний формат мозжечковой атаксии – заболевание Пьера-Мари. Преимущественно диагностируется в возрасте 25 лет и старше.
Мозжечковая атаксия бывает острая (на фоне вирусной инвазии, инфицирования), подострая, спровоцированная новообразованием, склерозом, хроническая, склонная к прогрессу, и пароксизмально-эпизодическая. Для уточнения, с какой формой необходимо бороться в конкретном случае, врач назначает анализы и исследования.
Симптомы атаксии
Стоит отметить, что каждая из форм заболевания проявляется по-разному. Между тем, для большинства патологий характерны следующие признаки:
-
нарушения координации движения рук, ног и тела;
Специфика проявления мозжечковой формы
Клиническая картина склонной к прогрессу формы болезни специфична, поэтому установить точный диагноз обычно легко. Предположить болезнь можно, исходя из общей симптоматики, поведения больного, принимаемым им позам. При наблюдении человека создается ощущение, что он пытается балансировать, для чего раскидывает руки в стороны. Больной стремится избегать поворотов туловищем, головой, произвольно падает от легкого толчка в момент попытки сдвинуть ноги, и даже не осознает этого. Конечности напряжены, походка сходна со свойственной пьяному, тело выпрямлено и откинуто назад.
По мере прогресса мозжечковая атаксия приводит к неспособности корректировать движения. Невозможно коснуться кончика носа. Меняется почек, речь, лицо похоже на маску, мышцы все время в тонусе, болит спина, шея, ноги и руки. Возможна судорожность, нистагм, косоглазие. У некоторых пациентов слабеет зрение, слух, становится сложно глотать.
Диагностика
Диагностика атаксии заключается, прежде всего, в выявлении ее формы. Для этого специалист осуществляет сбор анамнеза, проводит общий осмотр, отправляет больного на лабораторно-инструментальные исследования.
При сборе анамнеза врач спрашивает о перенесенных заболеваниях и употребляемых для их лечения лекарственных препаратах, наследственности. При общем осмотре он оценивает тонус мышц, качество зрения, слуха, рефлексы, а также проводит координационные пробы – пальценосовую и коленно-пяточную.
Кроме того, он может направить пациента на:
- общий и биохимический анализы крови и мочи – они позволяют исключить или подтвердить наличие сбоев в обмене веществ, а также выявить признаки отравления или воспаления;
- МРТ головного мозга – процедура помогает диагностировать атрофические процессы в верхней части отделов черепа;
- электроэнцефалографию – метод предназначен для оценки электрической активности отдельных участков головного мозга;
- компьютерную томографию головного и спинного мозга – результаты этого исследования помогают послойно посмотреть структуру тканей головного мозга, разглядев при этом возможные гнойнички, новообразования, кровоизлияния;
- спинномозговую пункцию с обязательным анализом ликвора;
- генетические тесты для выявления мутаций;
- диагностику ДНК, если имеют место наследственные патологии.
Важно! Для постановки точного диагноза крайне важно обратиться к опытному специалисту и пройти все необходимые обследования. В редких случаях патология может напоминать своей симптоматикой рассеянный склероз, тем самым, вводя в заблуждение медиков.
При регулярном приеме препарата значительно сокращается частота головокружений, уменьшается звон и шум в ушах, восстанавливается слух. Длительность терапевтического эффекта медикамента – от нескольких минут до 24 часов.
В сутки взрослому пациенту можно принять не более 48 мг бетагестина. Дозу препарата делят на 3 приема. Более точную дозировку определяет специалист в индивидуальном порядке.
Следует помнить и о том, что препарат способен вызвать побочные действия: тошноту, рвоту, диспепсию, вздутие живота, сильную головную боль, аллергические реакции.
Лечение атаксии
При выявлении инфекции применяется антибиотикотерапия. При рассеянном склерозе возможно использование гормональных препаратов и плазмафереза.
Обратите внимание! Залогом успеха лечения атаксии является своевременное выявление причины развития патологии и ее устранение. Именно поэтому опухоли удаляются хирургическим путем.
При диагностике отравления вводятся поддерживающие растворы. Дополнительно для укрепления организма врач может рекомендовать физические упражнения, направленные на повышение тонуса мышц. Облегчают состояние пациента также трости, ходунки или другие приспособления.
Опасности и вероятность заболеть
Преимущественный процент больных – лица, предрасположенные к атаксии в силу генетического фактора. С высокой долей вероятности понадобится лечение атаксии (мозжечковой, сенситивной или другого типа) лицам, перенесшим инфицирование мозга, страдающим эпилепсией и злокачественными новообразованиями. Выше вероятность атаксии при наличии пороков развития черепной коробки, мозга, а также проблемах кровотока.
Чтобы минимизировать рождение страдающих атаксией, при плохой наследственности необходимо очень ответственно подходить к вопросу продолжения рода. В ряде случаев врач может порекомендовать в принципе воздержаться от рождения детей. Это особенно актуально, если уже есть дети с атаксией, обусловленной генетикой.
Профилактика атаксии предполагает отказ от браков между близкими родственниками, терапию любых инфекционных очагов и контроль давления, а также соблюдение нормального распорядка дня, правильное питание. Следует избегать разновидностей спорта, занятие которыми сопряжено с риском ЧМТ.
Профилактика
Сложно предотвратить развитие наследственных патологий, между тем для снижения риска появления атаксии врачи рекомендуют:
- вести здоровый образ жизни, достаточно гуляя на свежем воздухе, занимаясь спортом и соблюдая режим дня и ночи;
- следить за своим здоровьем, регулярно посещать врача, вовремя лечить инфекционные заболевания в случае их выявления;
- следить за качеством питания;
- не откладывать с обращением к врачу в случае проявления похожих признаков.
Атаксии – патологии, которые могут диагностироваться в любом возрасте и серьезно влиять на качество жизни человека. Между тем, при всей своей коварности они поддаются корректировке, поэтому нуждаются в своевременном выявлении и лечении.
Чумаченко Ольга, врач, медицинский обозреватель
8, всего, сегодня
(190 голос., средний: 4,57 из 5)
В чем состоит терапия патологии?
Лечение мозжечковой атаксии предполагает обязательный прием следующих категорий медикаментов:
- миорелаксанты (помогают устранить спазм мышц);
- ноотропы (улучшают мозговую деятельность);
- препараты, которые помогают справиться с вестибулярными нарушениями;
- антидепрессанты;
- витамины группы B, витамин C и PP;
- противосудорожные лекарства.
Довольно часто мозжечковую атаксию сопровождает нистагм и дизартрия. Речь больного приобретает специфический характер: теряется плавность, наблюдается замедление и прерывистость. Также больной ставит ударение на каждый слог слов, в результате чего речь начинает напоминать скандирование. Иногда мозжечковая атаксия проявляется в результате мышечной гипотонии или сниженных глубоких рефлексов.
Важна своевременная диагностика поздней мозжечковой атаксии.
Организация лечебного процесса
Лечение заболевания организует невролог и оно в основном симптоматическое и включает в себя:
- Общеукрепляющее лечение — потребление витаминов группы В и других средств.
- Комплекс физических упражнений лечебной физкультуры, помогающих укрепить мышцы и нормализовать координацию движений.
Помимо описанного лечения для терапии атаксии требуется проведение коррекции иммунодефицита. Так, необходимо проведение курса приема иммуноглобулина. Лучевая терапия в этой ситуации противопоказана, а также нужно предотвращать чрезмерное облечение рентгеном и длительное нахождение под открытыми солнечными лучами.
Это важно! Важную роль в лечебном процессе могут играть медикаменты, восстанавливающие работу митохондрий, а именно рибофлавин, янтарная кислота, витамин Е.
Читайте также: