Агаммоглобулинемия Х-сцепленная инфантильная. Синдром Гарднера. Синдром Пейтца—Егерса.
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Синдром Гарднера - это наследственное заболевание, сопровождающееся полипозом толстого кишечника в сочетании с доброкачественными неоплазиями кожи, костей и мягких тканей. Может долгое время протекать бессимптомно. Возможны вздутие живота, урчание и расстройства стула. В некоторых случаях полипоз кишечника осложняется кровотечением или кишечной непроходимостью. Отмечается высокая вероятность развития колоректального рака. Заболевание диагностируется на основании жалоб, семейного анамнеза, данных осмотра, рентгенографии, КТ, МРТ, УЗИ, эндоскопии и других исследований. Лечение - эндоскопическая полипэктомия или резекция пораженных отделов кишечника.
МКБ-10
Общие сведения
Синдром Гарднера - редкая генетически обусловленная патология, при которой наблюдается диффузный полипоз толстого кишечника в сочетании с доброкачественными опухолями костей и мягких тканей (остеомами, фибромами, нейрофибромами, эпителиальными кистами и другими неоплазиями). Полипозом при синдроме Гарднера преимущественно поражаются прямая и сигмовидная кишка, однако полипы могут выявляться в других отделах кишечника. Впервые был описан американским врачом и генетиком Е. Дж. Гарднером в 1951 году. С тех пор в специальной литературе появились упоминания более чем о ста случаях данного заболевания. Риск малигнизации полипов толстой кишки с развитием колоректального рака в течение жизни составляет около 95%. Лечение проводят специалисты в сфере клинической проктологии, гастроэнтерологии, онкологии, ортопедии, стоматологии и челюстно-лицевой хирургии.
Причины
Синдром Гарднера передается по аутосомно-доминантному типу. Выраженность кишечных и внекишечных клинических проявлений может сильно варьировать. Первые симптомы синдрома Гарднера обычно появляются у детей старше 10 лет. Возможно позднее начало с образованием первых опухолей в возрасте старше 20 лет. В отдельных случаях наряду с полипозом толстого кишечника, остеомами и мягкотканными новообразованиями у больных синдромом Гарднера обнаруживаются полипы тонкого кишечника, желудка и двенадцатиперстной кишки.
Симптомы
Синдром Гарднера включает в себя характерную триаду: диффузный полипоз нижних отделов толстого кишечника, остеомы плоских и трубчатых костей, различные доброкачественные опухоли кожи и мягких тканей. При умеренном количестве и небольшом размере полипов кишечные проявления синдрома Гарднера могут отсутствовать или быть слабо выраженными. В подростковом или юношеском возрасте больные обычно впервые обращаются к врачам в связи с появлением доброкачественных костных и мягкотканных опухолей.
Остеомы при синдроме Гарднера могут локализоваться как в плоских, так и в трубчатых костях. Часто наблюдается поражение костей лицевого черепа, сопровождающееся обезображиванием. Возможно смещение и даже выпадение зубов. Через некоторое время после появления рост остеом у больных синдромом Гарднера прекращается, опухоли не озлокачествляются. Неоплазии мягких тканей отличаются большим разнообразием. Особенно часто выявляются липомы, дерматофибромы, нейрофибромы и эпителиальные кисты. Реже встречаются атеромы, лейомиомы и другие новообразования. Мягкотканные опухоли при синдроме Гарднера также протекают доброкачественно, малигнизация отсутствует.
Полипы толстой кишки при синдроме Гарднера нередко становятся случайной находкой при проведении исследований ЖКТ по другим поводам либо обнаруживаются в процессе расширенного обследования, назначенного в связи с появлением множественных мягкотканных и костных неоплазий. В течении синдрома Гарднера можно выделить три стадии поражения кишечника. На первой стадии заболевание протекает бессимптомно. На второй пациенты отмечают дискомфорт в животе, вздутие, урчание и периодические нарушения стула. В каловых массах могут обнаруживаться примеси крови и слизи.
На третьей стадии у больных синдромом Гарднера выявляются выраженный болевой синдром, постоянный метеоризм, обильные примеси слизи и крови в испражнениях, снижение веса, повышенная утомляемость, эмоциональная лабильность, нарушения электролитного и белкового обмена. У многих пациентов с синдромом Гарднера развивается анемия, обусловленная небольшими по объему, но часто повторяющимися кровотечениями из нижних отделов ЖКТ. В отдельных случаях у больных развиваются неотложные состояния, требующие экстренной медицинской помощи - обильные кишечные кровотечения или кишечная непроходимость.
Диагностика
Диагноз устанавливается на основании семейного анамнеза (наличия синдрома Гарднера у близких родственников), клинической картины, включающей в себя характерную триаду, и данных дополнительных исследований. При проведении физикального осмотра врач отмечает наличие множественных костных и мягкотканных опухолей различной локализации. У некоторых больных синдромом Гарднера выявляются деформации лица, обусловленные остеомами лицевого черепа. При пальпации костей туловища и конечностей могут обнаруживаться опухолевидные образования костной плотности. При поражениях легкой степени количество неоплазий может быть незначительным, что затрудняет диагностику.
При пальпации живота наблюдается болезненность в левой подвздошной области. На первой стадии поражения кишечника данный симптом может отсутствовать. При проведении пальцевого ректального исследования на слизистой прямой кишки больных синдромом Гарднера обнаруживаются множественные узлы. На контрастных рентгеновских снимках такие узлы отображаются в виде дефектов наполнения. При узлах небольшого размера (менее 1 см) информативность контрастного рентгенологического исследования снижается. В ходе ректороманоскопии выявляются полипы в прямой и ободочной кишке. Количество полипов может сильно варьировать.
У некоторых пациентов с синдромом Гарднера отмечаются ограниченные поражения отдельных участков кишки. В отличие от рентгенографии, эндоскопическое исследование дает возможность диагностировать полипы любого размера, в том числе - мелкие (диаметром от 1-2 мм). Для уточнения характера и распространенности костных опухолей при синдроме Гарднера осуществляют рентгенографию. При мягкотканных новообразованиях назначают КТ, МРТ или УЗИ области поражения. При необходимости выполняют биопсию полипов, остеом и мягкотканных новообразований.
Дифференциальную диагностику синдрома Гарднера врачи-проктологи проводят с обычными множественными полипами и другими формами семейного полипоза. Для разных вариантов наследственного полипоза характерны определенные отличия в преимущественной локализации полипов (поражение всего толстого кишечника, поражение дистальных отделов толстой кишки), характере патологических изменений костей и мягких тканей. Для уточнения этих различий перед постановкой окончательного диагноза проводят детальный внешний осмотр, осуществляют ирригоскопию и колоноскопию.
Лечение синдрома Гарднера
Лечение только хирургическое. Поскольку риск озлокачествления костных и мягкотканных неоплазий отсутствует, решение о проведении оперативных вмешательств принимают при наличии косметического или функционального дефекта. Полипоз толстого кишечника при синдроме Гарднера рассматривается, как облигатный предрак, поэтому многие врачи считают целесообразным проведение операции до появления признаков малигнизации. При небольшом количестве полипов возможна эндоскопическая полипэктомия.
При синдроме Гарднера с выраженным диффузным полипозом показана резекция пораженного участка кишечника или тотальная колэктомия с наложением илеостомы либо формированием илеоректального анастомоза (при отсутствии полипов прямой кишки). Хирургическое вмешательство рекомендуют проводить в возрасте 20-25 лет. Из-за калечащего характера операции молодые пациенты с синдромом Гарднера нередко отказываются от данного вмешательства. В подобных случаях показано динамическое наблюдение с проведением колоноскопии через каждые 6-8 месяцев.
Некоторые врачи являются сторонниками выжидательной тактики и считают, что колэктомию при синдроме Гарднера следует проводить только при появлении признаков озлокачествления или при часто повторяющихся кровотечениях с развитием анемии. Показанием к экстренному оперативному вмешательству при синдроме Гарднера являются обильное кишечное кровотечение и кишечная непроходимость.
Прогноз и профилактика
При своевременном адекватном лечении прогноз при синдроме Гарднера достаточно благоприятный. Тяжесть течения определяется выраженностью полипоза и локализацией внекишечных опухолей. Родителям, имеющим родственников, страдающих данным заболеванием, в период планирования беременности рекомендуют обратиться за медико-генетической консультацией.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
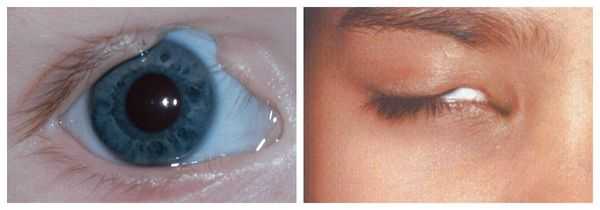
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
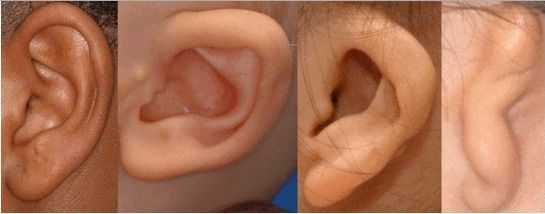
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
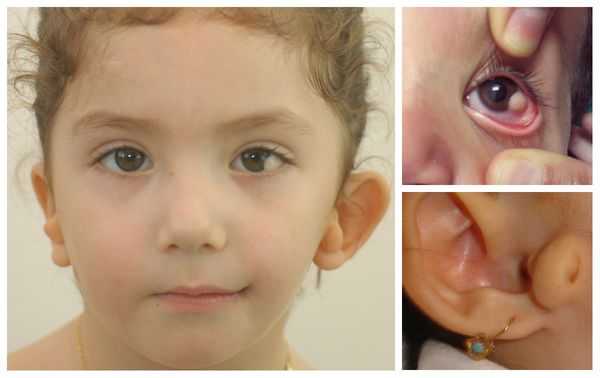
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
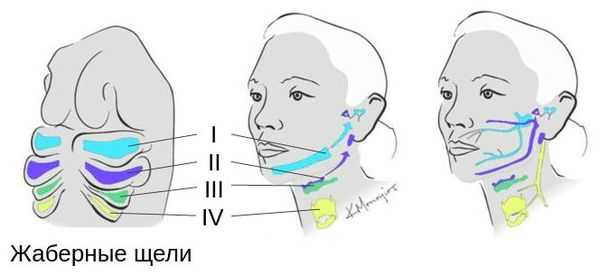
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
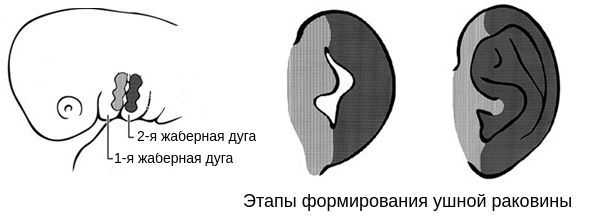
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
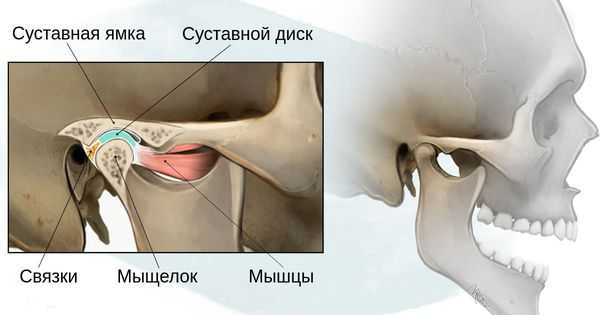
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
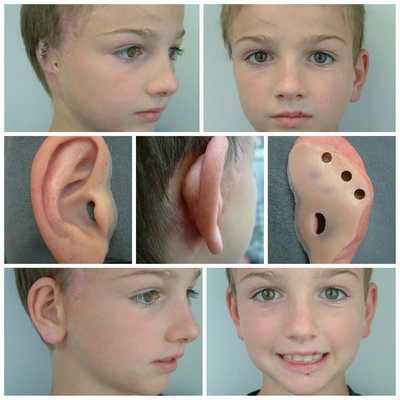
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Синдром Пейтца-Егерса ( Гамартомный полипоз )
Синдром Пейтца-Егерса — это редко встречающийся гамартомный гастроинтестинальный полипоз генетического происхождения. Проявляется абдоминальной болью разной интенсивности и локализации, метеоризмом, запорами, лентигообразной пигментацией, бледностью кожи и слизистых, частыми головокружениями и головной болью. Диагностируется с помощью УЗИ брюшной полости, контрастной рентгенографии органов ЖКТ, ЭГДС, колоноскопии и гистологического анализа биоптатов. Для терапии могут применяться ингибиторы циклооксигеназы-2, производные рапамицина, однако ведущими методами лечения являются лапаротомная или эндоскопическая полипэктомия, клиновидная резекция кишечника.
Гамартомный полипоз Пейтца-Егерса — редкое (орфанное) заболевание: ежегодно синдром регистрируется у 1 пациента на 25 000-300 000 населения. Одинаково часто выявляется у мужчин и женщин независимо от этнических или расовых отличий. Описание характерных признаков синдрома было впервые представлено в 1921 году в работах голландского врача Я. Пейтца и позднее дополнено в трудах американского доктора Г. Егерса. Заболевание чаще всего диагностируется в 30-40-летнем возрасте, нередко ассоциируется с опухолями ЖКТ, яичника, молочной железы и шейки матки. Полипы могут развиваться в любом отделе желудочно-кишечного тракта. Около 48% пациентов, страдающих гамартомным полипозом, умирают до 57 лет.
Синдром Пейтца-Егерса принадлежит к категории врожденных заболеваний, связанных с хромосомными аномалиями. Развитие гамартомного полипоза ЖКТ вызвано делецией, сплайсингом, инсерцией или другим вариантом мутации гена STK11, который расположен на коротком плече 19-й хромосомы в локусе 13.3. Поскольку явные изменения участка 19p13.3 определяются только у 70-80% больных с клиникой синдрома, специалисты в сфере клинической гастроэнтерологии и медицинской генетики допускают существование другого регуляторного гена, локализованного в позиции 19q13.4.
Болезнь Пейтца-Егерса передается по аутосомно-доминантному типу, однако наследственная отягощенность прослеживается у половины пациентов. Допускается, что в остальных случаях критическая мутация происходит в половых клетках родителей под воздействием различных повреждающих факторов (радиации, токсических химических веществ, вирусов, эндогенных свободных радикалов). Отличия в клинической картине синдрома, манифестировавшего у разных больных, по-видимому, связаны с особенностями экспрессии гена.
Патогенез
Механизм развития синдрома Пейтца-Егерса основан на несостоятельности серин-треониновой протеинкиназы 11, синтез которой кодируется мутировавшим геном STK11. За счет снижения функциональной активности энзима ускоряется пролиферация и клеточный рост, нарушаются межклеточные взаимодействия, угнетается апоптоз, что приводит к формированию полипов в желудочно-кишечном тракте. Ситуация усугубляется высоким риском малигнизации при наличии у пациента двух мутировавших аллелей или утрате гетерозиготности вследствие дополнительной соматической мутации.
В отличие от классических аденоматозных полипов, гамартомы у пациентов с болезнью Пейтца-Егерса отличаются нарушением нормального соотношения основных тканевых элементов желудочной или кишечной стенки. Гладкомышечные волокна собственной пластинки эпителиального слоя пролабируют в строму полипозного образования и разветвляются, создавая ложный эффект инвазии эпителия. При этом эпителиоциты имеют нормальную цитологическую структуру, отсутствуют признаки их активной пролиферации. В тонкокишечных полипах, кроме энтероцитов, выявляются бокаловидные клетки и клетки Панета.
Заболевание проявляется с рождения, у ребенка обнаруживают множественные лентиго — пигментные пятна темно-коричневого цвета размером от нескольких миллиметров до 1 см. При болезни Пейтца-Егерса они преимущественно располагаются на губах и слизистой оболочке ротовой полости, второй по частоте локализацией является промежность (на наружных половых органах и вокруг анального отверстия). Изредка пятна выявляют на нижних конечностях. У некоторых пациентов пигментация исчезает самостоятельно после периода полового созревания.
Другой характерный признак синдрома — боли в животе различной локализации и интенсивности. Болевые ощущения могут сочетаться с хроническими запорами и вздутием живота. Об осложненном течении синдрома Пейтца-Егерса свидетельствуют примеси крови в кале, иногда развиваются профузные кровотечения из ЖКТ. Наблюдаются нарушения общего состояния: бледность кожных покровов и слизистых оболочек, частые головные боли и головокружения, непереносимость физических нагрузок.
Осложнения
Вследствие постоянной мацерации поверхности полипов каловыми массами отмечаются хронические кровотечения различной интенсивности. При незначительной кровопотере формируется анемический синдром, о чем свидетельствует одышка, бледность, проявления сидеропении (изменение вкусовых пристрастий, ломкость волос и ногтей). В редких случаях при патологии Пейтца-Егерса возникают обильные кровотечения, представляющие опасность для жизни больного из-за быстрого развития коллаптоидного состояния.
Почти у 50% пациентов наблюдается инвагинация участка кишечника, которая проявляется очень сильными болями, может приводить к ишемии и некрозу кишки. Изменение нормальной структуры тонкого кишечника способствует появлению синдромов мальабсорбции и мальдигестии, сопровождающихся значительной потерей веса. Наиболее опасное осложнение — малигнизация полипов. Распространенность озлокачествления, по разным источникам, составляет от 10% до 24% всех случаев заболевания Пейтца-Егерса.
Постановка диагноза зачастую затруднена, что обусловлено низкой распространенностью синдрома Пейтца-Егерса и неспецифичностью диспепсических симптомов. Заподозрить заболевание можно при обнаружении у пациента выраженной пигментации, которая появляется уже в раннем возрасте, наличии данных о наследственной отягощенности. Диагностический поиск предполагает проведение комплексного обследования пищеварительной системы. Наиболее информативными являются:
- Сонография брюшной полости. Неинвазивное ультразвуковое сканирование применяется для экспресс-диагностики синдрома. УЗИ органов брюшной полости позволяет обнаружить объемные образования в просвете кишечника и патологию других органов ЖКТ. Сонография может быть недостаточно информативной при небольших размерах полипов (до 5 мм).
- Контрастная рентгенография. Выполнение рентгенограмм после перорального введения густой бариевой взвеси необходимо для выявления полиповидных разрастаний. При аномалии Пейтца-Егерса просматриваются дефекты наполнения округлой формы. Прогностически неблагоприятно обнаружение нечетких контуров или локального нарушения перистальтики.
- Эндоскопическое исследование. Для визуализации слизистой оболочки верхних отделов пищеварительной трубки осуществляют ЭГДС, для изучения состояния толстой кишки проводят колоноскопию. Полипы Пейтца-Егерса по внешнему виду напоминают аденомы, могут достигать нескольких сантиметров и иметь ножку. Иногда отмечают гиперемию и эрозии эпителия.
- Гистологический анализ. Цитоморфологическое изучение биоптатов необходимо для дифференцировки с другими новообразованиями. Для заболевания характерно сохранение всех слоев слизистой оболочки, отсутствие клеточной атипии и патологических митозов, разрастание стромы и гладкомышечных волокон. По тканевой структуре полипы являются гамартомами.
В общем анализе крови обнаруживают признаки анемического синдрома - уменьшение количества гемоглобина и числа эритроцитов. В биохимическом анализе крови выявляют снижение уровня общего белка. Всем пациентам выполняют копрограмму, для подтверждения кровотечений из ЖКТ проводят реакцию Грегерсена на скрытую кровь. По показаниям производят цитологический анализ кожных биоптатов из пигментированных участков.
Дифференциальный диагноз при симптомокомплексе Пейтца-Егерса в первую очередь осуществляют с другими формами полипоза (диффузным семейным, ювенильным). Основными диагностическими отличиями являются наличие гиперпигментации отдельных участков кожного покрова и специфическое гистологическое строение полипов. Также нужно дифференцировать заболевание с множественным лентиго. Для обследования пациента с признаками полипозного синдрома привлекаются специалист-гастроэнтеролог, проктолог, дерматолог.
Лечение синдрома Пейтца-Егерса
Этиопатогенетическая терапия не предложена. Из медикаментозных средств применяют нестероидные противовоспалительные препараты группы коксибов, которые ингибируют циклооксигеназу 2-го типа и замедляют развитие полипоза. В качестве потенциально эффективной терапии рассматривается назначение производных рапамицина, продемонстрировавших хорошие результаты при сочетании рака поджелудочной железы с патологией Пейтца-Егерса. Наиболее распространенным способом лечения является удаление образовавшихся полипов:
- При размерах новообразований до 15 мм: рекомендована эндоскопическая полипэктомия. Полипы желудка и двенадцатиперстной кишки удаляются в ходе ЭГДС, для иссечения полипов тонкого кишечника с учетом их локализации проводят интраоперационную или двойную баллонную энтероскопию.
- При полипах размерами больше 15 мм: показано лапаротомное хирургическое вмешательство с интраоперационной эндоскопической полипэктомией. Больным с множественным кишечным полипозом осуществляется классическая или лапароскопическая клиновидная резекция тонкого или толстого кишечника.
Исход заболевания зависит от распространенности полипоза и своевременности диагностических мероприятий. При диффузном поражении желудка и всех отделов кишечника прогноз относительно неблагоприятный, часто возникают кровотечения, возможна злокачественная трансформация полипов. Из-за врожденного характера синдрома меры специфической профилактики не разработаны. Для предупреждения осложнений необходимо активное динамическое наблюдение за пациентами с наследственным полипозом Пейтца-Егерса.
1. Синдром Пейтца-Егерса: обзор литературы и описание собственного клинического наблюдения/ Кайбышева В.О., Ивашкин В.Т., Баранская Е.К. и др.// РЖГГК - 2011 - №2.
2. Болезни поджелудочной железы, кишечника, системные заболевания с нарушением функций пищеварительного тракта/ Комаров Ф.И., Гребенев А.Л., Бурков С.Г. и др. - 1996.
3. Наследственные синдромы, ассоциированные с полипами и развитием злокачественных опухолей у детей/ Казубская Т.П., Белев Н.Ф., Козлова В.М. и др.// Онкопедиатрия — 2015 — Т.2, №4.
Синдром Пейтца-Егерса
Синдром Пейтца-Егерса (англ. Peutz-Jeghers syndrome) или гамартомный полипоз желудочно-кишечного тракта (ЖКТ) — орфанное (Запруднов А.М.) заболевание, характеризующееся множественными доброкачественными полипами (гамартомами) в желудке, тонкой и толстой кишке и, одновременно, пигментными пятнами тёмно-коричневого цвета, круглой или овальной формы, диаметром от 1 до 5 мм, располагающимися обычно на коже лица, вокруг рта и глаз, на носу, реже на конечностях, на слизистой оболочке полости рта, иногда гениталий и прямой кишки.
Синдром Пейтца-Егерса — заболевание, наследуемое по аутосомно-доминантному типу. Распространенность его, по данным разных авторов, составляет от 1 к 50 000 до 1 к 200 000 новорожденных. Частота заболевания одинакова для лиц мужского и женского полов.
Меланиновая пигментация губ и слизистой оболочки щёк при синдроме Пейтца-Егерса (Кайбышева В.О. и др.)
По Международной классификации болезней МКБ-10 синдром Пейтца-Егерса относится к «Классу XVII. Врожденные аномалии [пороки развития], деформации и хромосомные нарушения (Q00-Q99)», блоку «Q80-Q89 Другие врожденные аномалии», рубрике «Q85.8 Другие факоматозы, не классифицированные в других рубриках».
Полипы при синдроме Пейтца-Егерса обнаруживаются на протяжении всего желудочно-кишечного тракта: от желудка до прямой кишки. Обычно располагаются неравномерно: мелкие чаще локализуются в желудке, двенадцатиперстной и толстой кишке, а крупные — в тощей. Макроскопически полипы могут быть плоскими или высокими, различной величины (от нескольких миллиметров до 5 и более сантиметров), с гладкой либо дольчатой поверхностью, на широком основании и на ножке. Полипы располагаются по одиночке или кластерами, порой выстилая всю поверхность слизистой, напоминая ковровое покрытие. У пациентов с синдромом Пейтца-Егерса наряду с гамартомными полипами в толстой кишке и желудке часто обнаруживаются аденоматозные полипы (Кайбышева В.О. и др.).
Согласно теории эмбриональной дистопии полипы при синдроме Пейтца-Егерса рассматриваются как результат неправильного эмбрионального развития слизистой оболочки желудка. Они сохраняются в слизистой оболочке желудка с эмбрионального периода и обладают высокой потенциальной энергией роста (Эрдес С.И., Сергеева Т.Н.).
Синдром Пейтца-Егерса является так называемым «вероятно предраковым» заболеванием, с риском развития рака от 5 до 10 % (Тертычный А.С.).
Молекулярно-генетическая основа синдрома Пейтца-Егерса
Причина синдрома — герминальная мутация в генах LKB1 или STK11. Мутация этого гена приводит к сокращению длины белка и утрате киназной активности. Основные типы мутаций: небольшие делеции/инсерции, нонсенс, миссенс или большие делеции, приводящие к преждевременной терминации синтеза белка.
Мутации в гене STK11 определяются у 70-80% пациентов. Причина различий в диагностике мутаций бывает обусловлена методами, используемыми для их выявления. Кроме того, для пациентов с фенотипом синдрома, но без выявленных мутаций в гене STK11, обсуждается гетерогенность и возможность существования второго гена, ответственного за синдром в локусе 19q13.4 (Казубская Т.П. и др.).
Агаммоглобулинемия Х-сцепленная инфантильная. Синдром Гарднера. Синдром Пейтца—Егерса.
Заболевание встречается с частотой 1: 8 300-1:16 000 новорожденных.Тип наследования аутосомно-доминантный с различной степенью экспрессивности плейотропного гена, локализованного в локусе 5q21-q22.Плейотропный эффект гена объясняет симптоматику заболевания.
Первый постоянный признак синдрома, появляющийся в возрасте от 4 до 10 лет (редко позднее), - эпидермоидные кисты на коже лица, волосистой части головы, конечностей, реже груди, обычно развивающиеся задолго до полипоза толстой кишки, что облегчает раннее распознавание предракового заболевания. В дальнейшем они могут распространяться на туловище, промежность, конечности.Кисты существуют с рождения или появляются в раннем детстве, увеличиваясь в количестве до 20 лет.
Из других опухолей кожи часто обнаруживают трихоэпителиомы, кератоакантомы, липомы. Нередко развиваются фиброзные новообразования, особенно на месте травм, хирургического вмешательства, иногда встречаются фибросаркома, лейомиома желудка, кишечника.
Приблизительно у 50% больных выявляют костные изменения, главным образом в челюстных и клиновидных костях (остеомы, гиперостозы), развивающиеся в первой декаде жизни и имеющие доброкачественное течение. Размеры их небольшие, опухоли чаще множественные.
В прогностическом отношении наиболее важная составная часть синдрома - диффузный предраковый аденоматозный полипоз толстой кишки клинически проявляющийся обычно на втором десятилетии жизни и характеризующийся периодическими кровоточениями, вызывающими анемию и запоры.Более чем у 40% больных к 35-40 годам аденоматозный процесс в кишечнике завершается перерождением полипов в аденокарциному, повышен риск развития меланомы кожи и опухолей других органов - щитовидной железы, яичников, надпочечников, печени.
Диагноз ставят на основании характерной клинической картины, данных гистологического исследования, обнаружения полипов желудочно-кишечного тракта при систематическом инструментальном обследовании.
Диагностически значимы пигментные нарушения глазного дна (гиперплазия пигментного эпителия сетчатки).
Больных с множественными фиброзными опухолями кожи или пиломатриксомами рекомендовано обследовать для исключения синдрома Гарднера.
Гистологически эпидермальные кисты представляют собой полости, выстланные многослойным плоским эпителием, идентичным нормальному эпидермису. Содержимое кист образуют рыхлые слои кератина.
Дифференциальную диагностику проводят с другими опухолевыми синдромами, сопровождающимися поражением кожи и кишечника:
- болезнью Кауден;
- синдромом Пейтца-Егерса-Турена;
- синдромом Кронкайта-Канады;
- синдромом Мюира-Торре.
Опухоли кожи удаляют хирургическим путем или электрокоагуляцией после подтверждения диагноза. Основное лечение направлено на раннее профилактическое удаление полипов толстой кишки
Читайте также:
- Классификация курортов. Популярные советские курорты
- Инструменты в стоматологии. Стандартизация инструментов в стоматологии
- Диагностика нарушения подавления вестибулоокулярного рефлекса при головокружении. Методика
- Аптечки автомобилей. Требования к салону автомобилей
- Дружба и любовь. Влияние подруги