Апоптоз опухолевых клеток. Инкубация опухолевого материала.
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Цель. Изучить механизмы репарации повреждений ДНК и апоптоза в клеточных линиях гастроинтестинальных стромальных опухолей (ГИСТ) в результате воздействия на них некоторых групп химиопрепаратов in vitro для оценки перспектив проведения химиотерапии пациентам с неоперабельными и/или метастатическими формами ГИСТ, а также пациентам с развившейся резистентностью к таргетным препаратам, например, иматинибу. Материалы и методы. Проведено исследование чувствительности опухолевых клеток гастроинтестинальных стромальных опухолей (ГИСТ), к ингибиторам топоизомераз II типа (доксорубицину и этопозиду), препаратов, влияющих на динамическое состояние микротрубочек веретена деления винбластину, нокодазолу и паклитакселу), гидроксимочевине, цисплатине, а также таргетному препарату иматинибу. Экспрессию маркеров повреждения ДНК и апоптоза опухолевых клеток оценивали методом иммуноблоттинга. Результаты. Обнаружено, что клетки ГИСТ чувствительны к ингибиторам ДНК-топоизомеразы II типа. Вышеуказанные химиопрепараты индуцируют образование двунитевых разрывов ДНК в клетках ГИСТ и последующую их гибель по механизму апоптоза. Препараты, оказывающие влияние на веретено деления, а также цисплатин обладают наибольшим про-апоптогенным эффектом в отношении к клеткам ГИСТ. Выводы. Опухолевые клетки ГИСТ высоко чувствительны к препаратам, влияющим на динамическое состояние веретена деления, а также препаратам платины и, в меньшей степени, к ингибиторам топоизомераз II типа.
1. Dematteo R.P., Heinrich M.C., El-Rifai W.M., Demetri G . Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002; 33: 466-77.
2. Gramza A.W., Corless C.L., Heinrich M.C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15: 7510-8.
3. Liu Y., Tseng M., Perdreau S.A., et al. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 2007; 67: 2685-92.
4. McManus KJ, Hendzel MJ. ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol Biol Cell 2005; 16:5013-25.
5. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858-5868.
6. Tewari M, Quan LT, O'Rourke K, et.al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81(5):801-9.
7. Verweij J., Casali P.G., Zalcberg J., et al. Progression-free survival in gastrointestinal stromal tumors with high-dose imatinib: randomised trial. Lancet. 2004; 364: 1127-32.
В настоящее время проведение химиотерапии больным с ГИСТ считается нецелесообразным по причине имеющейся точки зрения о химиорезистентности данных злокачественных новообразований [1]. Поэтому основным нехирургическим методом лечения больных с неоперабельными, рецидивирующими и/или метастатическими формами ГИСТ является проведение им таргетной терапии иматинибом (Гливеком), воздействующей на активирующую мутацию тирозинкиназного рецептора с-KIТ в опухолевых клетках ГИСТ, следствием которой является усиление пролиферации и митотической активности опухолевых клеток ГИСТ. Однако, несмотря на высокую изначальную эффективность применения таргетных препаратов (в том числе, иматиниба) в лечении пациентов с ГИСТ с течением времени к ним начинает развиваться резистентность, существенным образом снижающая эффективность их дальнейшего применения. Например, было установлено, что после 2-х лет с момента начала проведения таргетной терапии иматинибом у более чем у половины пациентов с ГИСТ развивается резистентность к данному препарату, обусловленная развитием в клетках ГИСТ вторичных мутаций [2,7]. Несмотря на открытие и внедрение в практическую онкологию таргетных препаратов второго и третьего поколения (Сутент и Регорафениб, соответственно) высокая частота побочных эффектов от их применения, а также развитие к ним вторичной резистентности опухоли, являются стимулом для поиска новых альтернативных решений в терапии больных с неоперабельными, рецидивирующими и метастатическими формами ГИСТ.
Также известно, что существующее в настоящее время мнение о химиорезистености ГИСТ основано на результатах клинических наблюдений, проведенных до внедрения в практическую онкологию точных методов диагностики ГИСТ, позволяющих обнаружить совокупность гистологических и иммуногистохимических признаков, отличающих ГИСТ от других типов гладкомышечных и нейрогенных новообразований (например, лейомиосарком, лейомиом и шванном). Установлено, что данные типы опухолей обладают устойчивостью к большинству современных химиопрепаратов. Следовательно, отсутствие на момент проведения данных исследований точных методов дифференциальной диагностики ГИСТ и других гладкомышечных и нейрогенных злокачественных новообразований могли существенным образом повлиять на достоверность результатов исследований и привести к формированию ошибочного мнения о химиорезистентности ГИСТ.
В соответствии с вышеизложенным, представляло интерес изучение способности химиопрепаратов различных групп оказывать анти-пролиферативный эффект по отношению к клеткам ГИСТ и вызывать их гибель по механизму апоптоза.
Материалы и методы исследования. В качестве объекта исследования была выбрана клеточная линия ГИСТ Т-1, чувствительная к действию таргетного препарата иматиниба. Клетки культивировали в стандартных условиях (37оС, 5%СО2) в культуральной среде RPMI-1640 (ПанЭКО) с добавлением 10% эмбриональной телячьей сыворотки (HyClone), антибиотиков пенициллина-стрептомицина и L-глутамина (все реагенты ПанЭКО). Опухолевые клетки ГИСТ культивировали с доксорубицином, винбластином, нокодазолом, паклитакселом, иматинибом (Sigma), этопозидом, гидроксимочевиной (Calbiochem). Изменение уровня экспрессии белков-маркеров повреждения ДНК и маркеров апоптоза оценивали методом иммуноблоттинга с использованием соответствующих моноклональных антител (мАТ).
Результаты исследований и их обсуждение.
Было обнаружено, что инкубация опухолевых клеток ГИСТ Т-1 с химиопрепаратами индуцирует их гибель по механизму апоптоза. Об этом свидетельствовало значительное повышение уровней экспрессии расщепленных форм ПАРП и каспазы-3 в клетках ГИСТ после их инкубации с большинством из химиопрепаратов (РИС.1А). Наибольший про-апоптогенный эффект был обнаружен у препаратов платины, а также препаратов, влияющих на динамическое состояние микротрубочек веретена деления (в частности, винбластина). Ингибиторы топоизомеразы II типа доксорубицин и этопозид также вызывали гибель клеток ГИСТ по механизму апоптоза, тем не менее их эффект был менее выраженным. Примечательно, что уровень экспрессии одного из расщепленных фрагментов ПАРП (мол. массой 86 кДа) в опухолевых клетках ГИСТ коррелировал с уровнем экспрессии каспазы-3, что согласуется с данными литературы, свидетельствующими о способности каспаз (в том числе каспазы-3) индуцировать расщепление ПАРП на 2 фрагмента молекулярной массой 86 и 24 кДа, соответственно [6]. Важно отметить, что про-апоптогенный эффект ряда химиопрепаратов (этопозида, цисплатины, а также всех препаратов, влияющих на динамическое состояние микротрубочек) был более выраженным по сравнению с действием таргетного препарата иматиниба. Была также выявлена корреляционная зависимость между про-апоптогенным эффектом вышеназванных химиопрепаратов и их способностью индуцировать повышение уровня экспрессии гистона 2А, фосфорилированного по остаткам серина в положении 139 (-H2AX) (РИС.1Б). Известно, что данная форма гистона является общепризнанным маркером двунитевых разрывов ДНК, а также накапливается в М-фазе клеточного цикла, в частности, за счет ATM- или DNA-PKcs/Chk2-опосредованных механизмов фосфорилирования данной формы гистона даже на фоне отсутствия каких-либо повреждений ДНК, в том числе, двунитевых разрывов [4, 5]. В клетках ГИСТ, инкубированных с таргетным препаратом иматинибом, происходило умеренное повышение уровня экспрессии -H2AX, коррелировавшее с уровнем экспрессии расщепленной формы ПАРП, являющейся маркером апоптоза. Эти данные коррелируют с данными литературы, свидетельствующими о способности H2AX индуцировать апоптоз клеток ГИСТ под влиянием иматиниба [3].
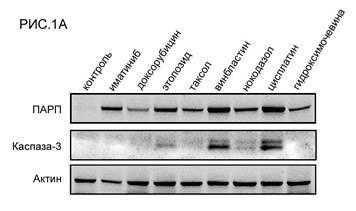
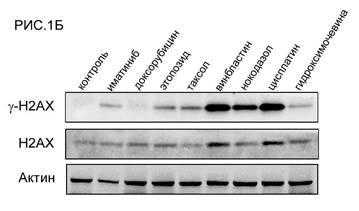
РИС. 1. Способность химиопрепаратов и таргетного препарата иматиниба индуцировать апоптоз (А) и вызывать фосфорилирование гистона H2AХ - -H2AX (Б) в клетках ГИСТ Т-1.
Таким образом, проведенные нами исследования свидетельствуют о способности химиопрепаратов индуцировать гибель клеток ГИСТ по механизму апоптоза. Наибольшим про-апоптогенным эффектом в отношении опухолевых клеток ГИСТ обладают препараты, влияющие на динамическое состояние микротрубочек, а также препараты платины. Полученные нами результаты свидетельствуют о чувствительности опухолевых клеток ГИСТ к некоторым химиопрепаратам, что ставит под сомнение существовавшую в настоящее время точку зрения о химиорезистентности у ГИСТ.
Заключение. Клетки ГИСТ чувствительны к ингибиторам топоизомеразы II типа, индуцирующим образование двунитевых разрывов ДНК и последующую гибель опухолевых клеток по механизму апоптоза. Наиболее эффективными химиопрепаратами в отношении клеток ГИСТ явились препараты, влияющие на динамическое состояние микротрубочек веретена деления - винбластин и паклитаксел. Полученные нами экспериментальные данные о чувствительности клеток ГИСТ к некоторым химиопрепаратам in vitro свидетельствуют о перспективности использования вышеназванных химиопрепаратов в лечении больных с неоперабельными, рецидивирующими и/или метастатическими формами ГИСТ, а также формами рефрактерными к монотерапии таргетным препаратом, в частности, иматинибом.
Работа финансировалась грантом Российского Научного Фонда (РНФ) № 14-15-00342.
Морфологическая и иммуногистохимическая характеристика почечно-клеточного рака
Маслякова Г.Н., Медведева А.В., Цмокалюк Е.Н., Воронина Е.С., Палатова Т.В.
Резюме
Ключевые слова
Статья
Введение. В последние десятилетия отмечается прогрессивное увеличение новых случаев возникновения злокачественных опухолей различных локализаций. Не является исключением и рак почки. В отличие от большинства зарубежных стран, где ежегодный прирост уровня заболеваемости почечно-клеточным раком (ПКР) составляет 2-3%, в России ежегодный прирост уровня заболеваемости ПКР составляет 6-10% [1,2,4,7,10,11,12,13]. Чаще ПКР растет медленно, имеет хорошо выраженную фиброзную капсулу, длительно не прорастает в окружающие почку ткани. Но с течением времени могут появиться признаки инвазии в окружающие почку структуры, а также в сосуды, что можно расценить как проявление агрессии раковой опухолью и опасным в плане метастазирования [3,5,6,12].
Часто злокачественные опухоли почки обнаруживают случайно при профилактическом осмотре, так как ПКР длительное время не дает клинических проявлений. При появлении жалоб у пациента с раковым заболеванием почки (боли в пояснице, повышение артериального давления, макрогематурия) уже обнаруживаются признаки инвазии опухоли в окружающие ткани и структуры почки или даже метастазирование [5,6,12]. Таким образом, актуальным остается задача раннего обнаружения и скорейшей точной диагностики опухолей в почках (локализации, степени дифференцировки и варианта ПКР).
Тактика ведения больных и прогноз выживаемости при злокачественных новообразованиях в почках зависят, прежде всего, от патологогистологического диагноза опухолевого процесса [5,6,8,9,12,14]. До сих пор гистологическое исследование при биопсии опухоли или удаленной опухоли почки является важнейшим в плане определения варианта ПКР, степени его дифференцировки и, соответственно, от окончательно поставленного патогистологического диагноза зависит дальнейшее лечение и прогноз для пациента. Важными прогностическими факторами при опухолях почек является степень ядерной атипии по Фурману, TNM стадия, клеточный вариант. По рекомендациям ВОЗ 2004 года существуют определенные гистологические признаки G (грейда или степени дифференцировки) почечно-клеточного рака: форма и размер ядер опухолевых клеток, распределение хроматина, форма и размер ядрышек, наличие митозов [7,12]. Чем выше дифференцировка опухоли (G1), тем более мелкие и однотипные ядра имеет опухоль (не более 10 мкм), хроматин и ядрышки не просматриваются. Со снижением дифференцировки (G3-G4), ядра становятся крупными, полиморфными, хроматин и ядрышки хорошо различимы [12]. Однако постановка диагноза рака почки и его дифференцировка по клеточной атипии бывает затруднена, особенно, если имеет место не классический светлоклеточный вариант, другие более редкие варианты (хромофобный, папиллярный, веретеноклеточный, мультилокуляный кистозный и другие). В таких случаях на помощь патологоанатому приходит иммуногистохимическое исследование.
Значимость иммуногистохимии опухолей трудно переоценить. В последние годы иммуногистохимическое исследование является обязательным для постановки окончательного патологогистологического диагноза в некоторых случаях, помогает максимально точно определить гистогенез опухоли, а также позволяет уточнить степень ее дифференцировки [4]. При изучении литературных источников мы не обнаружили данных, где в одном исследовании присутствовали бы результаты иммуногистохимического исследования наиболее распространенных вариантов ПКР с использованием большого количества антител (цитокератины, AMACR, Ki67, P53, PCNA, E-cadherin, VEGF, EGFR).
Цельисследования: выявление морфологических особенностей и определение наиболее важных иммуногистохимических маркеров нескольких вариантов ПКР для более точной и быстрой диагностики новообразований почек, что в дальнейшем позволит сформулировать прогноз для пациентов с опухолями в почках.
Материал и методы. Материалом для исследования явился послеоперационный материал 459 больных со злокачественными новообразованиями в почках эпителиального генеза (ПКР). Пациенты были прооперированы в клинической больнице им. Р.В. Миротворцева ФГБОУ ВО «Саратовский ГМУ им. В.И. Разумовского» Минздрава России в период с 2006 по 2011 год. Нами были проанализированы морфологические данные (локализация процесса, макрокартина, гистокартина) и дополнительно проведено иммуногистохимическое исследование цитокератинового профиля (СК 7, 8, 18), AMACR, Ki67, P53, PCNA, E-cadherin, VEGF, EGFR светлоклеточного, папиллярного, мультилокулярного кистозного, веретеоклеточного и хромофобного вариантов ПКР. Уровень экспрессии антител определялся путём суммарной оценки интенсивности окрашивания опухолевых клеток и числа позитивно окрашенных клеток. Случай расценивался как позитивный, если имело место любое по интенсивности окрашивание более 10 % опухолевых клеток. При окрашивании менее 10 % опухолевых клеток результат расценивался как нулевой. Слабая реакция более чем у 10 % опухолевых клеток со слабым окрашиванием оценивалась как «1+», при умеренном окрашивании более чем у 10 % опухолевых клеток - «2+», выраженное интенсивное окрашивание более чем 10% опухолевых клеток - «3+». В качестве группы сравнения выраженность экспрессии антител определялась в папиллярной аденоме (доброкачественная опухоль).
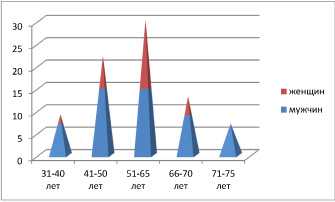
Результаты и обсуждение. По данным литературных источников, средний возраст, при котором обнаруживается ПКР, составляет 62 года [3,5,6,12]. Однако в нашем исследовании мы отметили, что средний возраст обнаружения рака почек 57 лет [8,9]. Некоторые случаи ПКР регистрировались в молодом и даже подростковом возрасте [14]. Кроме того, нами было отмечено, что у молодых пациентов с ПКР обнаруживаются редкие варианты опухолей почки (хромофобный, папиллярный, веретеноклеточный, мультилокулярный поликистозный). Также нами было отмечено, что редкие формы ПКР чаще встречаются у женщин [14], а у мужчин преобладал типичный светлоклеточный вариант.
Как известно из литературных источников [3,5,6,12], в последнее время все чаще стали выявляться злокачественные опухоли почек с признаками агрессии. Нами были отмечены признаки агрессии опухолей почек в 69,7% случаев (из 459 новообразований признаки инвазии имели 320). Естественно, что прогноз злокачественных опухолей любой локализации, в том числе и в почках, зависит от наличия или отсутствия метастазов, от стадии процесса и гистологического варианта заболевания, которые возможно дифференцировать только с помощью морфологического исследования [3,5,6,7,11,12]. Иногда опухоль имеет такое необычное строение или низкую дифференцировку, что постановка гистологического диагноза возможна только при использовании иммуногистохимического исследования [4]. Чтобы выявить эпителиальное происхождение опухоли (ПКР), мы использовали группу цитокератинов (СК, 7,8,18). Было обнаружено, что почти при всех вариантах ПКР СК18, дает более интенсивную реакцию (рис.1, рис.2), чем СК 7 и 8, кроме мультилокулярного кистозного варианта (табл.1). Доброкачественная опухоль почки (папиллярная аденома) показала одинаковую резко положительную реакцию на цитокератины СК7,8,18 (табл.1).
Каждый гистологический вариант ПКР показывал своеобразную комбинацию и интенсивность окрашивания как с цитокератинами, так и другими антителами (табл.1). Кроме цитокератинов, эпителиальный характер ПКР позволяют подтвердить следующие антитела: рецептор эпидермального фактора роста (EGFR) и AMACR. AMACR - специфический маркер для выявления рака предстательной железы, но он дает реакцию и на злокачественные эпителиальные опухоли других локализаций [4].
Дифференцировку (G - грейд) опухоли помогают установить антитела пролиферации и апоптоза. Р53, Ki-67, PCNA - маркеры пролиферации. Чем сильнее реакция маркеров пролиферации, тем более агрессивно ведет себя опухоль и соответственно имеет более низкую дифференцировку. Напротив, чем слабее реакция маркеров апоптоза ВАХ и Bcl-2, тем менее благоприятно для прогноза (рис.3). Маркер VEGF показывает васкуляризацию опухоли, чем сильнее реакция VEGF, тем лучше васкуляризирована опухоль и тем быстрее и агрессивнее она растет (рис.4).
Было отмечено, что папиллярный, хромофобный и муцинозный веретеноклеточный варианты ПКР имеют более агрессивное течение, чаще прорастают лоханку и капсулу почки, дают метастазы. Напротив, мультилокулярный кистозный вариант ПКР имеет более благоприятное течение и прогноз, никогда не метастазирует (рис.5, рис.6). Таким образом, для каждого варианта опухоли, также как и для степени дифференцировки (G) опухоли, имеется своеобразная комбинация реагирования и интенсивность окрашивания антител.
После проведения иммуногистохимических реакций в нашем исследовании была произведена коррекция диагноза для некоторых опухолей. Например, после проведения обычного гистологического исследования с помощью красителя гематоксилина/эозина в группе мультилокулярного поликистозного варианта ПКР насчитывалось 13 случаев. Как уже было сказано выше, этот вариант рака почки считается наиболее благоприятным - он чаще имеет высокий G грейд и никогда не дает метастазов [9]. В нашем исследовании в одном из 13 случаев мультилокулярного кистозного варианта после проведения иммуногистохимического исследования мы отметили такую же реакцию антител, как при светлоклеточном варианте. После дополнительного обследования у пациента обнаружены метастазы рака почки в лимфоузлы. Таким образом, вместо первоначально поставленного диагноза мультилокулярного поликистозного варианта ПКР был выставлен диагноз светлоклеточного ПКР с кистозной трансформацией, пациенту проведена коррекция лечения [9].
Заключение. Таким образом, в связи с увеличением частоты развития опухолей, их поздней диагностикой, агрессивности течения и неблагоприятного прогноза, морфологический метод исследования при опухолях почек является своевременным, необходимым и самым точным диагностическим методом. Также целесообразно использование дополнительно иммуногистохимического исследования при низкодифференцированных опухолях почки и может являться обязательным методом, последовательно выполняемым после стандартного гистологического исследования. Иммуногистохимический метод с помощью ряда антител позволяет провести дифференциальную диагностику различных вариантов ПКР, в некоторых случаях позволяет уточнить G (грейд) опухоли, а это позволяет быстро назначить правильное лечение и спрогнозировать течение заболевания.
Литература
- Давыдов М.И., Аксель Е.М. Злокачественные новообразования в России и странах СНГ в 2006 году / Под ред. М.И.Давыдова, Е.М.Аксель // Вестник Российского онкологического научного центра имени Н.Н. Блохина РАМН. - 2008. - Т. 19. № 2 (прил.1). - 152 с.
- Состояние онкологической помощи населению России в 2008 г. / Под ред. В.И. Чиссова, В.В. Старинского, Г.В. Петровой. М. ФГУ «МНИОИ им. П.А. Герцена Росмедтехнологий». 2009. 192 с.
- Клинико-морфологические и молекулярно-биологические особенности почечно-клеточного рака в прогнозировании результатов хирургического лечения / В.С. Дорошенко, А.Ф. Лазарев, С.Ф. Варламов и др. // Проблемы клинической медицины. - 2008. - № 1(13). - С. 39-48.
- Петров С.В., Райхлин Н.Т. (ред.) Руководство по иммуногистохимической диагностике опухолей человека. Издание 3-е, дополненное и переработанное. Казань. 2004. 451 с.
- Пономарева Ю.А. Клинико-морфологические критерии прогноза при раке почки. Автореф. дисс. … канд. мед. наук. С.-Пб. 2007. 19 с.
- Роль некоторых клинико-морфологических критериев в прогнозе рака почки / С.Х. Аль-Шукри, М.Г. Рыбакова, А.Э. Лукьянов, Ю.А. Пономарева // Ученые записки СПбГМУ им. акад. И.П. Павлова. - 2005. - Т.12. №3. С.34-38.
- Юрин А.Г. Опухоли почек (рабочие стандарты патологоанатомического исследования). - С.-Пб.: Издательство Санкт-Петербургского городского патологоанатомического бюро. 2006. 83 с.
- Целесообразность использования иммуногистохимического метода исследования для диагностики и прогнозирования течения низкодифференцированных случаев светлоклеточного варианта почечно-клеточного рак / Г.Н. Маслякова, А.В. Медведева, И.С. Аристова // Якутский медицинский журнал. - 2016. - 1(53). С.20-23.
- Клинико-морфологические особенности мультилокулярного кистозного варианта почечно-клеточного рака / Г.Н. Маслякова, А.В. Медведева, Е.Н. Цмокалюк, Т.В. Палатова // Вестник медицинского института «Реавиз». - 2016. - №2. С. 53-59.
- Automated uro-oncology data collection: the Cancer Research Uro-Oncology Database / Charlesworth PJ, Kilbey N, Taylor M, Leek R, Cranston D, Turner G, Crew J, Harris A, Protheroe A. // BJU Int. 2009. Nov. Vol. 20. [Epub ahead of print].
- Jemal A. Cancer statistics, 2002. Cancer J.Clin. 2002. Vol. 52. P. 23-47.
- Pathology and Genetics of Tumors of Urinary System and Male Genital Organs / Eds. John N. Eble, Guido Sauter, Jonathan I. Epstein, Esabell A. Sesterhen // World Health Organization Classification of Tumors. Lion, 2004. P. 5-76. 359 p.
- Smith R.A., Cokkinides V., Eyre H.J. American Cancer Society guidelines for the early detection of cancer, 2004. Cancer J. Clin. 2004. Vol. 54. P. 41-52.
- Occurrence of chromophobe renal cell carcinoma in a 15-year-old child (clinical case) / G.N. Maslyakova and A.V. Medvedeva // Journal of Case Report in Medical Science. 2016. 1(2). P.53-55.
Таблицы
Таблица 1. Сравнительная характеристика реагирования антител при иммуногистохимическом исследовании различных вариантов ПКР
VIII Международная студенческая научная конференция Студенческий научный форум - 2016

Апоптоз - программированная клеточная гибель, энергетически зависимый, генетически контролируемый процесс, который запускается специфическими сигналами и избавляет организм от ослабленных, ненужных или повреждённых клеток. Ежедневно, примерно около 5% клеток организма подвергаются апоптозу, а их место занимают новые клетки. В процессе апоптоза клетка исчезает бесследно в течение 15-120 минут.
Запрограммированная клеточная гибель это биохимически специфический тип гибели клетки, который характеризуется активацией нелизосомных эндогенных эндонуклеаз, которые расщепляют ядерную ДНК на маленькие фрагменты. Морфологически апоптоз проявляется гибелью единичных, беспорядочно расположенных клеток, что сопровождается формированием округлых, окруженных мембраной телец (“апоптотические тельца”), которые тут же фагоцитируются окружающими клетками.
Апоптоз - энергозависимый процесс, посредством которого удаляются нежелательные и дефектные клетки организма. Он играет большую роль в морфогенезе и является механизмом постоянного контроля размеров органов. При снижении апоптоза происходит накопление клеток, пример - опухолевый рост. При увеличении апоптоза наблюдается прогрессивное уменьшение количества клеток в ткани, пример - атрофия.
Морфологические проявления апоптоза.
Апоптоз имеет свои отличительные морфологические признаки, как на светооптическом, так и на ультраструктурном уровне. При окраске гематоксилином и эозином апоптоз определяется в единичных клетках или небольших группах клеток. Апоптотические клетки выглядят как округлые или овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина. Поскольку сжатие клетки и формирование апоптотических телец происходит быстро и также быстро они фагоцитируются, распадаются или выбрасываются в просвет органа, то на гистологических препаратах он обнаруживается в случаях его значительной выраженности. К тому же апоптоз - в отличие от некроза - никогда не сопровождается воспалительной реакцией, что также затрудняет его гистологическое выявление.
Наиболее четко морфологические признаки выявляются при электронной микроскопии. Для клетки, подвергающейся апоптозу характерно:
Сжатие клетки. Клетка уменьшается в размерах; цитоплазма уплотняется; органеллы, которые выглядят относительно нормальными, располагаются более компактно. Предполагается, что нарушение формы и объема клетки происходит в результате активации в апоптотических клетках трансглютаминазы. Этот фермент вызывает прогрессивное образование перекрестных связей в цитоплазматических белках, что приводит к формированию своеобразной оболочки под клеточной мембраной, подобно ороговевающим клеткам эпителия.
Конденсация хроматина. Это наиболее характерное проявление апоптоза. Хроматин конденсируется по периферии, под мембраной ядра, при этом образуются четко очерченные плотные массы различной формы и размеров. Ядро же может разрываться на два или несколько фрагментов. Механизм конденсации хроматина изучен достаточно хорошо. Он обусловлен расщеплением ядерной ДНК в местах, связывающих отдельные нуклеосомы, что приводит к развитию большого количества фрагментов, в которых число пар оснований делится на 180-200. При электрофорезе фрагменты дают характерную картину лестницы. Эта картина отличается от таковой при некрозе клеток, где длина фрагментов ДНК варьирует.
Формирование в цитоплазме полостей и апоптотических телец. В апоптотической клетке первоначально формируются глубокие впячивания поверхности с образованием полостей, что приводит к фрагментации клетки и формированию окруженных мембраной апоптотических телец, состоящих из цитоплазмы и плотно расположенных органелл, с или без фрагментов ядра.
Фагоцитоз апоптотических телец. Фагоцитоз апоптотических клеток или телец осуществляется окружающими здоровыми клетками, или паренхиматозными, или макрофагами. Апоптотические тельца быстро разрушаются в лизосомах, а окружающие клетки либо мигрируют, либо делятся, чтобы заполнить освободившееся после гибели клетки пространство. Фагоцитоз апоптотических телец макрофагами или другими клетками активируется рецепторами на этих клетках: они захватывают и поглощают апоптотические клетки. Один из таких рецепторов на макрофагах - рецептор витронектина, который является β3-интегрином и активирует фагоцитоз апоптотических нейтрофилов.
Участие апоптоза в физиологических и патологических процессах
Запрограммированном разрушении клеток во время эмбриогенеза (включая имплантацию, органогенез). Несмотря на то, что при эмбриогенезе апоптоз не всегда является отражением “запрограммированной смерти клетки”, это определение апоптоза широко используют различные исследователи.
Гормон-зависимой инволюции органов у взрослых, например, отторжение эндометрия во время менструального цикла, атрезии фолликулов в яичниках в менопаузе и регрессия молочной железы после прекращения лактации.
Удалении некоторых клеток при пролиферации клеточной популяции.
Гибели отдельных клеток в опухолях, в основном при ее регрессии, но также и в активно растущей опухоли.
Гибели клеток иммунной системы, как В -, так и Т-лимфоцитов, после истощения запасов цитокинов, а также гибели аутореактивных Т-клеток при развитии в тимусе.
Патологической атрофии гормон-зависимых органов, например, атрофии предстательной железы после кастрации и истощении лимфоцитов в тимусе при терапии глюкокортикоидами.
Патологической атрофии паренхиматозных органов после обтурации выводных протоков, что наблюдается в поджелудочной и слюнных железах, почках.
Гибели клеток, вызванных действием цитотоксических Т-клеток, например, при отторжении трансплантата и болезни “трансплантат против хозяина”.
Повреждении клеток при некоторых вирусных заболеваниях, например, при вирусном гепатите, когда фрагменты апоптотических клеток обнаруживаются в печени, как тельца Каунсильмана.
Гибели клеток при действии различных повреждающих факторов, которые способны вызвать некроз, но действующих в небольших дозах, например, при действии высокой температуры, ионизирующего излучения, противоопухолевых препаратов.
Биохимия апоптоза.
Активация цистеиновых (и некоторых других) протеаз — наиболее универсальная черта программируемой клеточной гибели независимо от организма, в котором она происходит. Основные участники программируемой клеточной гибели, каспазы («caspase» от «cysteine aspase») — это семейство эволюционно консервативных цистеиновых протеаз, которые специфически расщепляют белки по остаткам аспарагиновой кислоты. В настоящее время идентифицировано 10 каспаз. При апоптозе помимо активации цистеиновых протеаз, у растений выявлено возрастание активности сериновой и аспарагиновой протеаз.
Кроме того, в апоптозе принимают участие и другие протеазы, прежде всего, кальпаины, или Са2+-зависимые протеазы и убиквитин (протеаза, ковалентно связывающаяся с белком-мишенью). Эти протеазы — обязательный компонент каскада протеолитических ферментов. Так, ингибиторы кальпаина блокируют апоптоз. Убиквитин-протеосомный путь деградации белков активируется при апоптозе.
Роль каспаз в апоптозе разнообразна. Результатом активности протеаз являются характерные изменения в морфологии клеток при апоптозе.1. Гидролиз белков ламинов, армирующих ядерную мембрану. Это ведет к распаду ядерной оболочки и конденсации хроматина. Мишенями протеаз при апоптозе являются также белки ядрышек, гистоны и негистоновые белки и топоизомераза. Топоизомераза — связующее звено между ДНК хроматина и белковыми структурами ядра, с помощью которого хроматин прикрепляется к ядерному матриксу. Расщепление топоизомеразы — это этап образования высокомолекулярных фрагментов ДНК.
2. Расщепление антиапоптозных белков — протеолиз ингибитора ДНКазы, ответственной за фрагментацию ДНК. В нормальных клетках апоптозная ДНКаза CAD (caspase-activated DNase) образует неактивный комплекс с ингибитором 1CMiwm DFF (DNA fragmentation factor). При апоптозе ингибитор Гмс участием каспаз 3 и 7 инактивируется и свободная CAD, вызывая нуклеосомные разрывы хроматина, ведет к образованию фрагментов ДНК с молекулярной массой кратной молекулярной массе ДНК в нуклеосомных частицах — 180-200 пар нуклеотидов. Эти фрагменты и дают характерную лесенку ДНК при электрофоретическом разделении в агарозном геле. Апоптоз возможен и без фрагментации ДНК. Обнаружен ядерный белок ACCINVS (apoptotic chromatin condensation inducer in the nucleus), который при комбинированном действии каспазы 3 и неидентифицированной протеазы расщепляется на фрагменты. Один из них в присутствии дополнительных неядерных факторов вызывает апоптотическую конденсацию хроматина и фрагментацию ядра (кариорексис) без фрагментации ДНК. Кроме непосредственной активации нуклеаз, протеазы (путем ограниченного протеолиза) устраняют структурное разобщение между нуклеазами и ДНК в составе хроматина, удаляют белки, защищающие ДНК.3. Угнетение репарации ДНК: инактивирование и нарушение регуляции белка, участвующего в репарации ДНК, а также в сплайсинге мРНК, репликации ДНК. Мишенью каспаз является поли-(АДФ-рибозо)-полимераза (ПАРП), которая участвует в репарации ДНК (катализирует полиАДФ-рибозилирование белков, связанных с ДНК). Донором АДФ-рибозы является NAD'. Активность ПАРП-полимеразы возрастает в 500 раз и более при связывании с участками разрыва ДНК. ПАРП участвует в репарации поврежденной ДНК, регуляции активности эндонуклеаз, поддержании структуры хроматина посредством АДФ-рибозилирования. Апоптотическая гибель клетки сопровождается расщеплением ПАРП каспазами. При массированных разрывах ДНК чрезмерная активация ПАРП, сильно снижая содержание внутриклеточного NAD*, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по пути некроза.4. Разрушение белков цитоскелета. Деградация структурных и функциональных белков митотического аппарата.5. Участие в экспрессии генов. Эта функция связана с протеолизом репрессоров и с образованием пептидов, регулирующих транскрипцию (модификация факторов транскрипции). Субстратом протеаз является, например, гистон, выступающий репрессором генов.6. Одна из функций протеаз — передача апоптозного сигнала от индукторов апоптоза. Сигналы могут быть трансмембранными, рецептор-зависимыми. Рецепторами служат трансмембранные белки. Протеазы принимают участие либо непосредственно при взаимодействии индукторов апоптоза с рецепторами, либо через активацию протеинкиназ, играющих важную роль в передаче трансмембранного сигнала с целого ряда рецепторов.Локализация протеаз в различных отделах (компартментах) клетки способствует эффективной трансмембранной передаче сигналов программируемой клеточной гибели. Часть протеаз связаны с мембранами (цитоплазматической, ядерной, мембранами органелл или вакуоли) — это мембраносвязанные протеазы. Другие — находятся в матриксе ядра, цитоплазмы или органелл. Аспарагиновая протеаза растений, по всей видимости, локализована в вакуоли. Предполагается, что сериновые протеазы локализуются в цитоплазме и в ядре. Известно, что в ядрах протеазы могут быть прочно ассоциированы с хроматином и, в том числе, непосредственнно с гистонами. Перемещение протеаз в клетке может сопровождаться их активацией. Например, повышение концентрации Ca2+ внутри клетки способствует перемещению Са2+-зависимой протеазы и протеинкиназы из цитоплазмы в мембрану. При этом происходит автокаталитическая активация неактивных форм протеазы.Так, активация некоторых протеаз может быть обусловлена увеличением концентрации кальция в клетках, наблюдаемой при разных типах апоптоза (раздел выше). АФК также могут быть непосредственными индукторами активации протеаз. Появление локальных участков однонитевой ДНК активирует, например, ядерные ДНК-зависимые сериновые протеазы, специфичные к гистону.
Множество ветвей сигнальной трансдукции перепроверяет правильность выбранного алгоритма событий на пути к апоптозу, уберегая клетку от бессмысленной гибели. Выявлено несколько механизмов, ограждающих клетку от случайного самоуничтожения с участием протеаз.
Во-первых, протеазы синтезируются в клетке в неактивной форме, а процессинг неактивных форм протеаз происходит путем автолиза или путем протеолиза другими протеазами. Например, каспазы синтезируются в клетке в виде прокаспазы — неактивного мономера с молекулярной массой 30-50 кДа. Активные формы — тетрамеры, содержащие по две субъединицы: (р 10 — р20)2 (рис. 9.7). Прокаспазы обладают незначительной протеолитической активностью, составляющей 1-2% активности зрелой каспазы. Механизм протеолитического само- или перекрестного расщепления (ауто- или транс-процессинга), а затем пространственного сближения ведет к образованию активных каспаз. От прокаспазы отделяется регуляторный N-концевой домен (продомен), а оставшаяся часть молекулы разделяется на большую (около 20 кДа) и малую (около 10 кДа) субъединицы. Затем происходит ассоциация большой и малой субъединиц. Два гетеродимера образуют тетрамер с двумя каталитическими центрами, работающими независимо. Первоначально концентрация каспаз в клетке ничтожна. Благодаря свойству автокатализа, концентрация активных каспаз может возрастать лавинообразно.Во-вторых, протеазы обратимо взаимодействуют с эндогенными белковыми ингибиторами, образуя неактивные комплексы (латентные комплексы описаны для цистеиновых, Са2+-зависимых и некоторых других протеаз). При действии различных индукторов апоптоза происходит диссоциация неактивных комплексов протеаза-ингибитор. Обратимое взаимодействие Са2+-зависимых протеаз с эндогенными ингибиторами регулируется кальцием. Цистеиновая протеаза связывается ковалентно с ингибитором через дисульфидную связь. Высвобождение и активация каспазы происходит в результате тиол-дисульфидного обмена и сопряжена с окислительно-восстановительным состоянием клетки и метаболизмом глюкозы.В-третьих, протеазы могут быть компонентами специальных рецептор-зависимых систем. Так, [рецептор + лиганд + адаптер + прокаспаза] формируют специфический агрегат, в котором происходит активация каспаз. Такой агрегат называют апоптосомой или апоптозным шапероном. Самое интересное, что выявлены консервативные области гомологии (в том числе NB-область) белка адаптера у животных и продуктов генов резистентности у растений, включая томат, арабидопсис и табак. Более того, белки похожи структурно. Предполагается, что продукты гена резистентности могут играть роль адаптеров в апоптосоме. Таким образом, при узнавании продукта авирулентности, по всей видимости, происходит диссоциация апоптосомы и развертывание программы апоптоза.
Продукты генов резистентности, по-видимому, ответственны за эффективность гибели клеток при заражении — узнавание факторов и запуск машины самоуничтожения, за первые (самые важные) шаги на пути к стремительной гибели клетки.Существует несколько путей реализации программы ПКГ. Путь передачи сигнала: индукторы — рецепторы — адаптеры — каспазы первого эшелона — регуляторы — каспазы второго эшелона. Рецептор взаимодействует с лигандом. Насколько обратима гибель клетки? На этапе активации каспаз первого эшелона жизнь клетки еще можно сохранить. Существуют регуляторы, которые блокируют или, напротив, усиливают разрушительное действие каспаз первого эшелона. После активации каспазами первого эшелона каспаз второго эшелона путем протеолиза из прокаспаз процесс, запушенный программой смерти, становится необратим. Эти каспазы способны в дальнейшем к самоактивации (автокатализу или автопроцессингу) и активируют фактор фрагментации ДНК на нуклеосомные фрагменты. Вернемся к митохондриям. Апоптотическое изменение митохондрии может индуцироваться окислительным стрессом, повышением концентрации Ca2+. При апоптозе из межмембранного пространства митохондрий высвобождаются белки — апоптогенные факторы:
AIF (Apoptosis Inducing Factor) — флавопротеин с молекулярной массой 57 кДа. Будучи добавлен к изолированным ядрам, он вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям — высвобождение цитохрома С и каспазы 9. Высвобождаемый цитохром С вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) образует комплекс с прокаспазой. APAF-I играет роль арматуры, на которой происходит аугокаталитический процессинг каспазы 9 (мультимерная арматура APAF1-цитохром-С-комплексов напоминает пропеллер). Обнаружены ингибиторы высвобождения цитохрома С, блокирующие апоптоз, например, белок Bel.
Список используемой литературы:
Гордеева А.В., Лабас Ю.А., Звягильская Р.А.Апоптоз одноклеточных организмов: механизмы и эволюция Биохимия, 2004, том 69, вып. 10, с. 1301—1313
Голубев А.М., Москалева Е. Ю., Северин С.Е., Веснянко Т.П., Кузовлев А.Н., Алкадарский А.С., Порошенко Г.Г. Апоптоз при критических состояниях
Апоптоз опухолевых клеток. Инкубация опухолевого материала.
Апоптоз представляет собой систему контроля клеточной дифференцировки, обеспечивающую самоуничтожение дефектных структур. Тем самым поддерживая нормальное функционирование целостного организма. Регуляторами апоптоза являются белки семейства Bcl-2, которые могут обладать как антиапоптическими свойствами, так и проапоптическими. Проапоптические белки запускают синтез цитохрома С в миохондриях, провоцирующего образование апоптосомы, активирующей каспазы, которые обладают способностью к денатурации белков, что приводит к гибели клетки. Для обнаружения дефектов в клетке существует белок р53, который при наличии повреждений в клетке активирует синтез проапоптических белков. При недостаточном апоптозе, дефектные клетки пролиферируют, что приводит к их злокачественной трансформации. Злокачественные клетки подвергаются серии генетических изменений. Если это способствует их преимущественному росту над нормальными клетками, то риск развития новообразований значительно возрастает. Потеря предшественника апоптоза - белка P53 является наиболее распространенным механизмом уклонения от гибели. Канцерогенез можно рассматривать как сложный клеточный процесс, который связан с неограниченным репликативным потенциалом, независимостью от сигналов роста и параллельным сопротивлением ингибирующему рост сигналу, уклонение от активации клеточной смерти, устойчивый ангиогенез, а также способность тканевой инвазии и метастазирования.
1. Кузнецов, С.Л. Гистология, цитология и эмбриология / С.Л. Кузнецов, Н.Н. Мушкамбаров Н. - М.: МИА, 2007.
2. Галицкий В.А. Возникновение эукариотических клеток и происхождение апоптоза // Цитология, 2008, том 47, вып. 2, с. 103-120
3. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70. 10.1016/S0092-8674(00)81683-9.
4. Taylor, R. C., Cullen, S. P., and Martin, S. J. (2008). Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231-241.
5. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49-63.
7. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015.
8. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 2013; 5: a008656.
9. Manzl C, Fava LL, Krumschnabel G, Peintner L, Tanzer MC, Soratroi C et al. Death of p53-defective cells triggered by forced mitotic entry in the presence of DNA damage is not uniquely dependent on Caspase-2 or the PIDDosome. Cell Death Dis 2013; 4: e942.
10. Shalini S, Dorstyn L, Dawar S and Kumar S: Old, new and emerging functions of caspases. Cell Death Differ. 22:526-539. 2015.
Клеточный гомеостаз в организме здорового человека определяется балансом между гибелью и пролиферацией клеток. Дефекты, возникающие в процессах дефференцировки и новообразования клеток, ведут к самоуничтожению этих структур [2]. Может показаться парадоксальным, что стимуляция клеточной гибели может способствовать выживанию организма.
Механизм, отвечающий за инициирование и выполнение запрограммированной гибели клеток, называется апоптозом. Он осуществляется под действием внеклеточных или внутриклеточных факторов. Под воздействием этого процесса, ДНК распадается на фрагменты, клетка сжимается, клеточные мембраны разрушаются, происходит элиминация и она поглощается соседней клеткой или специфичной клеткой имунной системы. Особенностью этого процесса является то, что мембрана клетки не разрушается до полного завершения этапов самопроизвольной гибели. Что дает возможность избежать риска возникновения воспалительных процессов. Обычно от начала запуска апоптоза до окончательной клеточной фрагментации требуется несколько часов. Однако этот период зависит от типа клетки, стимула и апоптотического пути [1,2].
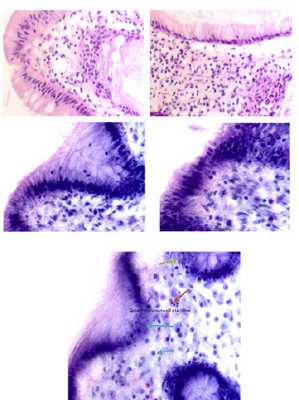
Апоптозные клетки выглядят как округлые либо овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина [1]. (Рис. 1.).
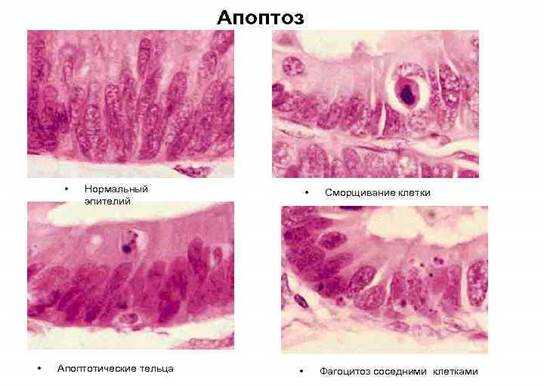
Рис. 1. Стадии апоптоза эпителиальной клетки
Основными регуляторами запрограммированной гибели клеток являются белки, принадлежащие к семейству Bcl-2. Эти белки могут, как активировать апоптоз, то есть быть проапоптотическими, так и ингибировать его, обладая антиапоптотическими свойствами. Антиапоптотические белки в здоровой клетке связывают и инактивируют проапоптотические. Это происходит тогда, когда она не нуждается в гибели [4,5].
Существует два основных пути апоптоза: внутренний и внешний. Внешний путь обеспечивает связывание «лиганд смерти» с «рецептором смерти». Внутренний путь контролируется митохондриями и выделением цитохрома С. Регуляторы апоптоза взаимодействуют со специфическими рецепторами на мембране клетки, называющимися «рецепторы смерти», которые связываются с молекулами, сигнализирующими гибель, как часть внешнего апоптотического пути. Это связывание вызывает эффект апоптоза [7].
Контроль такой элиминации производится в митохондриях, которые обеспечивают внутреннюю часть апоптического пути. В этих органеллах содержатся сигнальные молекулы, связаные с митохондриальной мембраной и известные как цитохром C. В ответ на проапоптотические сигналы из проапоптотических белков, высокую концентрацию Ca 2+ в цитозоле или гипоксию цитохром C высвобождается в клетку митохондриями и связывается с белком. Это приводит к образованию апоптосомы. После образования апоптосома активирует группу белков под названием каспазы, которые денатурируют другие белки в клетках [8]. Так как активные каспазы могут разрушающе воздействовать внутри здоровой клетки, они производятся в неактивной форме - прокаспазы. В фазе апоптоза семейство каспаз представляет собой основные эффекторные молекулы самого процесса элиминации, которые вносят вклад в конечные стадии апоптотической гибели клеток путем компонентов цитоскелетного аппарата и ядерной ДНК [4,9]. Все каспазы подразделяются на инициаторы, эффекторы и стимуляторы. Инициаторы расщепляют и активируют каспазы эффекторы, амплифицируя сигнал. Эффекторы расщепляют различные белки, что приводит к процессу апоптоза. Активация каспаз ведет к запуску протеолитического каскада реакций, провоцирующих гибель клетки [9].
Помимо каспаз, чрезвычайно важным является белок P53, обеспечивая обнаружение повреждения ДНК, аномалий хромосом и остановку клеточного цикла. Если повреждения необратимые, то апоптоз актимируется. P53 активирует процесс путем увеличения продуцирования проапоптотического белка, который активирует каспазный каскад, что в конечном итоге приводит к самоуничтожению клетки [10].
Апоптоз — это защита организма от персистенции пораженных клеток, которые могут оказаться потенциально опасными для многоклеточного организма. Однако, эти процессы могут нарушаться, сопровождаясь либо чрезмерным апоптозом, например, в случае дегенеративных заболеваний, либо недостаточным, что приводит к ускоренной пролиферации дефектных клеток и возникновению онкологии [2]. Проблемы недостаточного самоуничтожения клеток могут возникать на любом этапе апоптоза, что приводит к злокачественной трансформации пораженных клеток, метастазированию опухолей и устойчивости к противоопухолевым препаратам [6]. Следовательно, устойчивость к апоптозу или угнетение этого процесса играют жизненно важную роль в канцерогенезе. Одной из причин торможения процессов самопроизвольной гибели клеток является нарушения баланса проапоптотических и антиапоптотических белков. Проапоптотические (например, Bax, Bad, Bcl-Xs) и антиапоптотические белки (например, Bcl-2, Bcl-XL, Mcl-L и т. Д.) являются двумя основными группами белков семейства Bcl -2. Антиапоптотические белки регулируют апоптоз, блокируя выделение цитохрома С в митохондриях, в то время как проапоптические -стимулируют . Нарушение этого баланса вызывает угнетение процесса апоптоза в пораженных клетках. Причиной может стать как чрезмерная экспрессия антиапоптотических белков, так и проапоптотических или их комбинации.
Так, в основе возникновения В-клеточной лимфомы лежит механизм, подавляющий синтез проапоптического белка семейства Bcl-2, что приводит к торможению апоптоза клеток фолликулярной лимфомы и их пролиферацию [3]. (Рис. 2.).
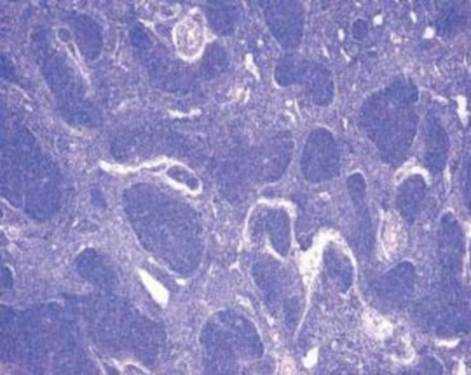
Рис. 2. Гистопрепарат ткани лимфатического узла. Фолликулярная лимфома.
Новообразования можно рассматривать как результат последовательности генетических изменений, в течение которых нормальная клетка превращается в злокачественную. И именно несостоявшийся апоптоз таких клеток является одним из существенных критериев, которые вызывают злокачественную трансформацию [3]. Злокачественные клетки подвергаются серии генетических изменений. Если это способствует их преимущественному росту над нормальными клетками, то риск развития и роста новообразований значительно возрастает. Например, когда в клетках кожи возникают повреждения под воздействием ультрафиолетового излучения (например, солнцем, соляриями), обычно срабатывает апоптоз. Это помогает устранить патологические элементы. Если апоптоз не происходит, такие клетки могут выживать и пролифирировать, превращаясь в злокачественные. (Рис.3.).
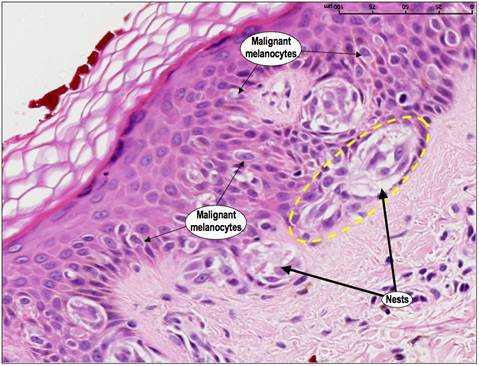
Запрограммированная гибель также играет роль в распространении онкологического процесса. Чтобы злокачественная клетка переместилась в другую часть тела ( метастазирование), она должна уметь выживать в крови или лимфатических системах и проникать в окружающие ткани. Апоптоз предотвращает эти процессы [6]. Наиболее распространенным механизмом уклонения от гибели является потеря предшественника апоптоза - белка P53 или его мутация. Более половины всех онкологических пациентов имеют мутированный или отсутствующий ген, программирующий синтез р53, что приводит к повреждению или отсутствию белка. При некоторых опухолевых процессах р53 трансформируется из активатора апоптоза в ингибитор, тем самым он не только не индуцирует апоптоз, а его блокирует. Это же провоцирует метастазирование новообразований, так как позволяет обходиться злокачественным клеткам без специфического микроокружения, избегая самопроизвольной гибели [3,9]. Однако, ингибирование процессов апоптоза не единственное условие для роста и развития новообразований. Без устойчивого ангиогенеза, самодостаточности сигналов роста и нечувствительности к сигналам против роста, «прото-раковые» клетки все еще могут умирать другими способами, даже если они избегают апоптотической запрограммированной клеточной смерти [6].
Выводы. Таким образом, канцерогенез можно рассматривать как сложный клеточный процесс, который связан с неограниченным репликативным потенциалом, независимостью от сигналов роста и параллельным сопротивлением ингибирующему рост сигналу, уклонение от активации клеточной смерти, устойчивый ангиогенез, а также способность тканевой инвазии и метастазирования. Злокачественные опухоли являются инвазивными и могут метастазировать в отдаленные места через систему кровообращения. Следовательно, метастатическое распространение, а не первичная опухолевая нагрузка, является основной причиной смертей от рака [6,4,7].
Читайте также:
- Снохождения и ночной энурез. Ночные страхи как причина эпилепсии
- КТ, МРТ при кисте второй жаберной щели
- Врожденный нефротический синдром
- Шагание на месте. Чесательный рефлекс и рефлексы мышечного спазма
- Температура роста бактерий. Мезофильные бактерии. Термофильные бактерии. Психрофильные бактерии. Аэрация бактерий.