Болезнь Альцгеймера. Ослабление памяти при амилоидных бляшках
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
В народе болезнь Альцгеймера часто называют старческим маразмом. С каждым годом количество заболевших людей неуклонно растет. Отчасти это связано с увеличением продолжительности жизни. Первые признаки заболевания малозаметны, человек испытывает проблемы с запоминанием новой информации. Сначала страдает кратковременная память, но с прогрессированием болезни возникают проблемы с долговременной памятью, нарушается речь и когнитивные функции.
Болезнь может не проявлять себя на протяжении нескольких лет. После обращения за профессиональной помощью и обнаружения заболевания пациенты, в среднем, живут около 7 лет. Своевременная диагностика и грамотно спланированная терапия способна продлить жизнь больного на 12-15 лет.
Причины развития деменции альцгеймеровского типа до конца не понятны. Известны лишь ключевые моменты, к которым относится формирование бляшек из бета-амилоида и скопление нейрофибриллярных клубков в разных отделах головного мозга. Современная медицина способа для остановки или замедления дегенеративных процессов не имеет - действия специалистов будут направлены на смягчение симптомов и облегчение состояния больного.
Причины развития и прогрессирования болезни Альцгеймера
Многие исследователи пытаются выяснить, почему с возрастом в тканях мозга человека начинаются дегенеративные процессы. Существует три гипотезы, объясняющие возможные причины развития данного заболевания:
- тау-гипотеза является одной из самых молодых, находящейся в стадии изучения. Согласно этой теории, патологические процессы в головном мозгу, приводящие к развитию деменции, запускаются при изменении структуры тау-белка. Сначала внутри нервных клеток образуются нейрофибриллярные клубки, вызывающие сбой в работе транспортной системы нейрона. Затем прекращается передача сигналов между отдельными клетками, после чего наступает и гибель нейронов;
- амилоидная гипотеза была открыта сравнительно недавно - ей еще нет 30 лет. В качестве основной причины развития дегенеративных процессов были названы скопления бета-амилоида. В поддержку этой теории было проведено много различных исследований, но вакцина, способная очистить мозг от белковых бляшек, также не является эффективной в ходе лечения деменции. Несмотря на то, что амилоидная теория научным сообществом принята в качестве основной, она не дает возможность объяснить все действия, происходящие в мозгу человека при развитии болезни Альцгеймера. Достоверно известно, что большое количество амилоидных бляшек не является непосредственной причиной заболевания - они лишь приводят к запуску дегенеративных операций. Что же действительно является пусковым механизмом к началу отложения бета-амилоида, пока не известно;
- холинергическая гипотеза была принята сразу после того, как началось изучение проблемы снижения умственных возможностей у пожилых людей. Ее авторы считали, что патология развивается в результате снижения синтеза ацетилхолина, являющегося нейромедиатором. С течением времени это предположение стало считаться маловероятным, так как медикаментозное снижение дефицита ацетилхолина малоэффективно при лечении деменции альцгеймеровского типа. Тем не менее, на основе этой теории было разработано большое количество методов поддерживающей терапии, многие из которых успешно используются и в нынешнее время.
Кроме того, многие исследователи считают, что к развитию болезни Альцгеймера может привести чрезмерное употребление сахара в пищу. Глюкоза блокирует выработку энзима, который необходим для предотвращения развития дегенеративных процессов в тканях головного мозга.
Описание заболевания
О том, что в старости происходит ослабевание рассудка, писали еще древнегреческие и римские врачи. Но впервые на эту болезнь обратил внимание Алоис Альцгеймер, который в 1901 году начал наблюдение за Августой Детер. Ей было 50 лет, и ее слабоумие с каждым днем прогрессировало. После смерти пациентки, врач опубликовал материалы, содержащие анализ заболевания, названного в последствии его именем.
Изначально болезнью Альцгеймера именовали деменцию, развивающуюся у молодых пациентов, тогда как людей преклонного возраста относили к категории лиц, у которых развивается сенильная деменция. На сегодняшний день диагноз деменция альцгеймеровского типа ставится при наличии характерных симптомов, которые развиваются определенным образом и сопровождаются типичными признаками нейропатологии. Возраст пациента при этом значения не имеет.
Болезнь Альцгеймера принято делить на четыре основных стадии, на каждой из которых отмечается прогресс нарушений функций:
- Предеменция. Признаки, характерные для данного этапа заболевания, часто путают с реакцией организма на стрессовые ситуации. Небольшие нарушения когнитивных функций начинают проявляться примерно за 5-7 лет до того, как будет поставлен точный диагноз. Выявить отклонения можно лишь при проведении детального нейрокогнитивного тестирования. Появляются первые проблемы с памятью, пациент испытывает трудности с усвоением нового материала. Также утрачивается способность к планированию, появляются первые проблемы с концентрацией внимания и абстрактным мышлением, но они мало заметны на данном этапе развития заболевания. Нередко появляется апатия, считающаяся наиболее устойчивым признаком деменции на всех ее стадиях.
- Ранняя деменция чаще характеризуется нарушениями речи и основных исполнительных функций. Расстройства памяти на некоторое время отступают на второй план. На этом этапе человек еще способен оперировать простыми понятиями в процессе речевого общения, выполняет повседневные задачи без посторонней помощи, прекрасно помнит события из прошлого или заученную когда-то информацию. Но со стороны больной может казаться неловким - это связано с наличием некоторых координационных проблем и утратой способности к планированию движений своего тела.
- Умеренная деменция. Прогрессируя, болезнь приводит к постепенной утрате способности к самостоятельной деятельности. Расстройства становятся очевидными, пациент утрачивает доступ к собственному словарному запасу. Он произносит неправильные слова, вставляя их в речь вместо забытых. Координация нарушается настолько, что больной утрачивает способность к выполнению большинства повседневных задач. На этом этапе заметно ухудшаются все виды памяти, человек уже не узнает членов семьи и других близких людей.
- Тяжелая деменция является последней стадией развития болезни. Человек на этом этапе уже не в состоянии обходиться без помощи других людей. Речевое общение сводится к произношению отдельных фраз или слов, постепенно и эта способность утрачиваетс . Резко снижается масса тела, больной пребывает в состоянии сильной апатии.
Итогом деменции альцгеймеровского типа становится гибель пациента. Однако истиной причиной летального исхода нередко становятся сторонние заболевания и патологии, такие как пневмония или пролежневые язвы.
Диагностика болезни
Перед постановкой диагноза врач изучает историю его болезни, беседует с родственниками, проводит собственные наблюдения. Для выявления характерных признаков болезни и исключения других заболеваний, имеющих схожую картину, применяются аппаратные методики, в том числе, компьютерная и магнитно-резонансная томография.
На ранних стадиях врачи для диагностики используются различные нейропсихологические тесты, поскольку неврологический осмотр, как правило, отклонений не выявляет. Поэтому исследование должно быть более обширным. При оценке состояния больного проводится беседа с его родственниками, поскольку сам пациент не способен заметить нарушений и больным себя не считает.
По результатам анализа крови выявляются причины деменции, которые не связаны с прогрессированием болезни Альцгеймера. Устранение их может способствовать тому, что симптомы заболевания будут устранены.
Сравнительно новыми способами диагностики заболевания являются методики, позволяющие наблюдать бета-амилоидные скопления в тканях головного мозга при помощи ПЭТ-сканера. О развитии заболевания также можно судить по количеству тау-белка или бета-амилоида в биологической жидкости из спинного мозга пациента.
Как правило, диагностика на ранних этапах болезни Альцгеймера проводится редко. Прежде всего, это связано с тем, что ни сам больной, ни его родственники не замечают отклонений от нормы и не обращаются за квалифицированной помощью. Диагноз ставится уже тогда, когда функциональные нарушения становятся слишком явными.
Как уже отмечалось выше, пациенты, в конечном итоге, погибают не от самой болезни Альцгеймера, а от сопутствующих заболеваний. Продолжительность жизни больного зависит от разных факторов, но исследователи отмечают, что в среднем женщины живут дольше мужчин.
Терапия болезни Альцгеймера и уход за больным
Как не печально это осознавать, но на сегодняшний день лекарства от болезни Альцгеймера не существуют. Ни одно из средств, применяемых при терапии этого заболевания, не способно прекратить или приостановить развитие деменции. Исследователи ведут работу в этой области, на крысах и мышах тестируются новые лекарства, но в современной медицине применяются лишь препараты, способные устранить некоторые из симптомов.
При работе с пациентом, у которого начинает развиваться деменция альцгеймеровского типа, часто используют различные психологические подходы:
- поведенческая терапия поможет определить предпосылки и последствия проблемного поведения, а также провести их коррекцию. Данный метод не способен оказать существенного влияния на общее состояние больного, но смягчает некоторые из проблем;
- эмоциональная терапия помогает больным с легкими формами болезни адаптироваться к своему состоянию. Работа проводится индивидуально или в группах, пациенты осуждают памятные события из прошлого, рассматривают фотографии или видеофильмы;
- когнитивная терапия использует методы, позволяющие пациентам осознать окружающую обстановку, а также понять собственное место в ней. Когнитивная переподготовка позволяет добиться улучшения состояния больного, но с прогрессированием заболевания эффект исчезает;
- стимулирующие методы включают в себя терапии, направленные на общее укрепление организма, а также создание положительного эмоционального фона. Арт-терапия, музыкальная терапия, общение с животными и занятия физкультурой позволяют несколько улучшить повседневную жизнь людей, страдающих болезнью Альцгеймера.
Поскольку заболевания является неизлечимым и носит дегенеративный характер, то членам семьи больного приходится организовать постоянный уход и присмотр за ним. Это является неотъемлемой частью терапии на протяжении всей болезни.
На заключительных стадиях болезни основной задачей врачей и родственников пациента становится облегчение его состояния. На этом этапе развиваются сопутствующие заболевания, требующие профессионального вмешательства.
Профилактика развития заболевания
Исследования, касающиеся причин развития болезни Альцгеймера, не дают точных указаний к тому, как предотвратить деменцию. Но, все же, существуют некоторые некоторые факторы, способные оказать влияние на состояние здоровья человека в преклонном возрасте:
- Рацион питания. Было выявлено, что люди, живущие в Средиземноморье, реже страдают болезнью Альцгеймера. В частности, это связано с особенностями местного питания. Оливковое масло, красное вино, рыба и морепродукты способны оказать смягчающее влияние на это заболевание. Полезны также крупяные культуры, пшеница, овощи и фрукты. Для мозга необходим куркумин, который содержится в популярной специи. Также есть доказательства пользы каприловой кислоты, входящей в состав кокосового масла. Употребление этого продукта позволяет пациентам вернуть способность выполнять простые домашние дела.
- Высокий уровень холестерина, диабет, табакокурение, а также другие факторы развития заболеваний сердца и всей сердечно-сосудистой системы, нередко ассоциируются с риском развития тяжелых форм болезни Альцгеймера.
- Тренировка мозга и интеллектуальные занятия. Замечено, что люди, увлекающиеся чтением, разгадыванием ребусов и кроссвордов, играющие на музыкальных инструментах и владеющие иностранными языками, заболевали болезнью Альцгеймера в более позднем возрасте. Интеллектуальные нагрузки отодвигают время наступления возрастных патологических изменений в тканях головного мозга.
Также некоторые исследователи обратили внимание на взаимосвязь риска развития болезни с условиями работы пациента. Считается, что магнитные поля могут спровоцировать наступление болезни Альцгеймера. Среди внешних факторов также называются тяжелые металлы и растворители, попавшие в организм. Но достоверных данных о том, как внешняя среда воздействует на процесс развития дегенеративных процессов, на сегодняшний день нет.
Болезнь Альцгеймера для большинства государств является серьезной проблемой, так как деменция накладывает большую нагрузку и ответственность на общество. Это заболевание может развиться у любого человека, независимо от его статуса и материального положения. Наиболее обсуждаемым случаем было заболевание Рональда Рейгана, бывшего президента Соединенных Штатов Америки. Факт его болезни был освещен многими средствами массовой информации и послужил поводом для написания научных работ, рассматривающих нарушение когнитивных функций у известных людей, имеющим незаурядные способности и острый ум.
Болезнь Альцгеймера. Ослабление памяти при амилоидных бляшках
Российская медицинская академия последипломного образования
Российская медицинская академия последипломного образования, Москва
25 лет амилоидной гипотезе происхождения болезни Альцгеймера: достижения, неудачи и новые перспективы
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2016;116(6‑2): 3‑9
Амилоидная гипотеза развития болезни Альцгеймера (БА) продолжает доминировать, однако за свою 25-летнюю историю концепция значительно изменилась. Оказалось, что накопление β-амилоида связано не только с повышением его продукции (как казалось после открытия генетических механизмов части семейных случаев БА), но и с нарушением его клиренса из ткани головного мозга, который осуществляется при помощи микроциркуляторной системы. При этом наиболее патогенную роль в веществе мозга играют не сами сенильные бляшки, описанные А. Альцгеймером почти 110 лет назад и состоящие из нерастворимых конъюгатов, а растворимые олигомеры β-амилоида. Связь сосудистого и дегенеративного процессов при БА подтверждают единство факторов риска, а также клинические, нейровизуализационные, патоморфологические и экспериментальные данные. Одним из звеньев, связующих дегенеративный и сосудистый процессы при БА, служит инсулинорезистентность. Исходя из современного состояния амилоидной гипотезы обсуждаются вопросы новых мультимодальных стратегий терапии БА.
Идея, что накопление β-амилоида может быть одним из основных механизмов развития болезни Альцгеймера (БА), была впервые выдвинута в 1984 г. G. Glenner и соавт. [2], но не нашла последователей и вызвала много критики. Лишь в 1991 г. J. Hardy и соавт., обнаружив в одном из семейных случаев БА мутацию гена белка-предшественника амилоида на 21-й хромосоме, нашли неопровержимое доказательство справедливости амилоидной гипотезы патогенеза БА, которая таким образом стала ведущей не только для объяснения причин развития заболевания, но также для разработки подходов к диагностике и поиску новых терапевтических стратегий [3, 4]. Последующие подтверждения амилоидной гипотезы были связаны с выявлением в других семейных случаях БА мутаций генов пресенилина-1 и -2, которые также приводили к гиперпродукции β-амилоида.
β-Амилоид - белок, состоящий из 40-42 аминокислот и образующийся при протеолитическом расщеплении трансмембранного белка - предшественника амилоида (англ.: amyloid precursor protein - APP). В норме APP расщепляется ферментом α-секретазой с образованием растворимого α-APP, который выводится из мозга, и пептида, состоящего из 83 аминокислот, который остается в мембране. В последующем мембранный пептид под действием второго фермента - γ-секретазы, локализованной внутри трансмембранной зоны, расщепляется на два небольших пептида - p7 и p3, которые не являются «амилоидогенными». При генетически детерминированных формах БА показано, что при заболевании нарушается процесс расщепления APP. В результате на первом этапе расщеплению c помощью β-секретазы подвергается внеклеточная часть APP, вследствие чего образуются растворимый β-APP и остающийся в мембране пептид, состоящий не из 83, а из 91 аминокислоты, а затем под действием γ-секретазы образуется патологический β-амилоид (A β) 1-42, который накапливается в мозге.
За 25 лет своей истории амилоидная теория претерпела ряд ключевых изменений. Отложение в межклеточном пространстве агрегатов β-амилоида с формированием сенильных бляшек длительное время рассматривалось в качестве основного фактора развития заболевания. К настоящему моменту стало понятным, что патогенное действие β-амилоида заключается не только и не столько в образовании амилоидных бляшек, состоящих из нерастворимых агрегатов амилоида, сколько в токсическом эффекте растворимых олигомеров амилоидного белка, которые запускают целый каскад повреждающих механизмов, включающих нейровоспалительный процесс, окислительный стресс, эксайтотоксичность, которые в конечном итоге приводят к утрате синапсов, связей между нейронами и их гибели. При этом снижение численности синапсов может отражать утрату способности нейронов поддерживать функционирование аксонов и дендритов либо гибель нейронов.
Еще недавно считалось, что первоначально (на преклинической стадии) патологический процесс связан с накоплением β-амилоида, которое, достигнув критического уровня, нарушает метаболизм τ-протеина, с накоплением которого в форме интранейрональных нейрофибриллярных клубочков и их «экспансией» в веществе мозга коррелируют клинические проявления заболевания. В настоящее время показано, что по крайней мере в значительной части случаев дегенеративный процесс начинается с нарушения метаболизма и агрегации τ-протеина либо параллельной конверсии β-амилоида и τ-протеина в растворимые олигомерные формы, которые взаимно усиливают токсичность друг друга и последовательно поражают различные отделы мозга, возможно, благодаря механизмам, напоминающим распространение патологического прионного белка.
Кроме того, оказалось, что накопление β-амилоида связано не столько с изменением структуры и избыточной продукцией нерастворимых фрагментов белка, а преимущественно с нарушением выведения β-амилоида из мозга. При этом весьма обоснованным выглядит предположение, что β-амилоид является элементом нормальной работы ЦНС, отвечающим за формирование памяти, нейрогенез, и в ограниченных количествах присутствует у любого здорового человека.
β-амилоид и сосудистые факторы риска
В последние годы накоплены данные, свидетельствующие об условности границ между дегенеративным и сосудистым процессами в развитии деменции. По данным крупных эпидемиологических исследований, артериальная гипертензия, курение, сахарный диабет, гиперлипидемия обусловливают повышение риска как БА, так и сосудистой деменции [5, 6]. Тесная связь сосудистого и дегенеративного процессов представляет огромный интерес, так как воздействуя на модифицируемые факторы риска, можно влиять не только на частоту сосудистых заболеваний, но и БА 7.
Сопутствующая сосудистая патология служит своего рода «катализатором» клинической манифестации БА и способствует более раннему дебюту деменции [16]. В ряде исследований показана связь между альцгеймеровскими изменениями и атеросклерозом крупных экстра- и интракраниальных артерий 17, но не патологией мелких сосудов 21. Тем не менее проспективно было показано, что прирост количества амилоидных бляшек связан с уменьшением эластичности сосудистой стенки и ареактивностью сосудов [23].
Атеросклеротическое повреждение крупных сосудов, снижение эластичности стенки малых сосудов приводят к гипоперфузии вещества мозга и при определенных условиях (генетически детерминированных) способствуют запуску патофизиологического каскада, приводящего к развитию БА. В условиях гипоксии ткани происходит более быстое формирование β-амилоида из АРР в связи с нарушением регуляции активности ферментов β- и γ-секретазы, обеспечивающих метаболизм данного белка 24. Важная роль гипоперфузии в отложении β-амилоида подтверждена на экспериментальных моделях. Так, при двустороннем пережатии сонной артерии у крыс в исходе отмечены накопление β-амилоида в лептоменингеальных артериях и увеличение числа корковых микроинфарктов 28.
В то же время накопление амилоида в межклеточном пространстве и стенке сосудов приводит к увеличению сосудистого сопротивления, нарушению эндотелийзависимой вазодилатации и/или индукции вазоконстрикции, снижению перфузионного кровотока в покое [30, 31]. Таким образом формируется «порочный круг»: гипоперфузия и гипоксия могут индуцировать отложение β-амилоида, что в свою очередь может способствовать усугублению ишемического повреждения.
Накопление β-амилоида в мозге - результат гиперпродукции или нарушения его клиренса?
После обнаружения роли мутаций генов АРР, пресенилина 1 и 2 основным механизмом накопления β-амилоида считалось повышение его продукции. Однако в настоящее время, особенно при позднем начале заболевания, накопление амилоида связывают с нарушением его выведения из ткани головного мозга. Амилоид, наряду с другими белками и растворимыми метаболитами с интерстициальной жидкостью может выводиться из мозга по периваскулярным пространствам (пространства Вирхова-Робина), в конечном итоге - в шейные лимфатические узлы.
Учитывая, что направление движения по периваскулярным пространствам противоположно внутрисосудистому току крови, механизм работы данной дренажной системы остается не до конца понятным. При математическом моделировании данной «транспортной» системы стало понятно, что продвижение по периваскулярному пространству осуществляется прерывисто, за счет обратной рефлекторной волны, с максимальной скоростью, сразу после прохождения пульсовой волны по сосуду. Еще одной особенностью периваскулярного транспорта веществ является необходимость создания однонаправленного клапанного механизма: за счет веществ, способных тесно прилегать к сосудистой стенке, чем создается препятствие обратному току жидкости и достигается более поступательное движение в направлении, обратном пульсовой волне.
Соответственно при атеросклерозе проксимальных церебральных сосудов, снижении реактивности и эластичности их микроциркуляторного русла, увеличении жесткости сосудистой стенки происходят не только нарушение прямого тока крови к мозговому веществу за счет снижение интенсивности пульсовой волны, но и нарушение обратного периваскулярного дренажа, что приводит к формированию зоны гипоперфузии и накоплению в этих же участках продуктов обмена и жизнедеятельности клеток.
Снижение амплитуды движений сосудистой стенки может способствовать накоплению β-амилоида как непосредственно в ткани мозга, так и в периваскулярном пространстве с последующей инфильтрацией сосудистой стенки в виде амилоидной ангиопатии. Это обусловливает необходимость поиска новых подходов для сохранения функционирования данной системы с помощью препаратов, способных влиять на свойства эндотелия и эластичность сосудистой стенки.
Врожденная иммунная система и β-амилоид
Врожденная иммунная защита ЦНС осуществляется благодаря активному участию микроглии. В процессе развития БА микроглия может играть двоякую роль. На раннем этапе функция микроглии скорее защитная: активация фагоцитоза может способствовать дополнительной элиминации амилоида из ткани, активизации трофических факторов с развитием репаративных процессов. Но со временем активизация микроглии оборачивается повышением продукции провоспалительных цитокинов: IL-1β, TNF-α, STAT3, IL-6, IL-12, IL-23, что влечет за собой дополнительное повреждение нервной ткани, а также замедляет репаративные процессы.
Амилоидные бляшки
Амилоидные бляшки представляют собой внеклеточные отложения β-амилоида, содержащие также убиквитин, пресенилин 1 и 2, аполипопротеин и другие белки. β-Амилоид образуется вследствие нарушения естественного расщепления трансмембранного АРР, что приводит к накоплению патологического β-амилоида во внеклеточном пространстве с формированием амилоидных бляшек (см. рисунок).
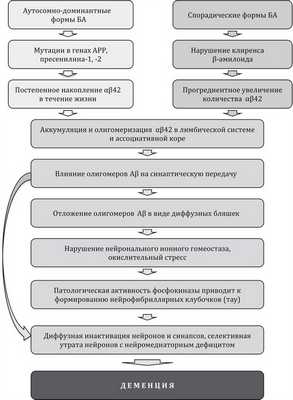
Патогенез БА (наследственные и идиопатические формы).
Центральная часть бляшки действительно представляет собой конгломерат из нерастворимых цепей белка, «ядро» бляшки окружено пенумброй, которая представляет собой скопление растворимых изомеров белков. Показано, что β-амилоид начинает откладываться за 25 лет до появления первых симптомов болезни. Но к моменту появления когнитивного снижения накопление β-амилоида останавливается и именно с этого момента наиболее активно запускаются дегенеративные процессы с активным прогрессированием деменции. Благодаря этому феномену выдвинули гипотезу о «протективном» эффекте β-амилоида, однако это сугубо гипотетическое предположение.
β-амилоид и инсулинорезистентность
Инсулин, наряду с участием в углеводном, жировом и белковом обмене, является сигнальной молекулой в головном мозге. Уровень инсулина в ЦСЖ превышает его плазматический уровень в десятки и сотни раз. Инсулин в головном мозге является модулятором синаптической пластичности, играя важную роль в процессах обучения, обеспечивая процессы консолидации и извлечения следов памяти.
Наибольшая концентрация инсулиновых рецепторов в головном мозге наблюдается в гиппокампе и миндалине. Кроме того, инсулин выполняет нейротрофическую функцию за счет активизации сигнального пути PI3K/AKT (фосфоинозид-3-киназы/протеинкиназы), который обеспечивает рост нейронов, защиту клеток мозга от апоптоза, их пролиферацию и метаболизм. У больных БА обнаружено снижение активности протеина IRS-1 (субстрат инсулинового рецептора), участвующего в активации данного сигнального механизма, что способствует нарушению инсулиновой регуляции и формированию инсулинорезистентности. В условиях недостатка глюкозы происходит изменение активности ферментов в сторону гиперфосфорилирования тау-протеина, что способствует формированию нейрофибриллярных клубочков, а также инактивации киназы гликогенсинтетазы-3 (GSK-3), что в свою очередь приводит к нарушению укладки и агрегации β-амилоида. Кроме того, инсулин регулирует активность холинацетилтрансферазы, обеспечивает синтез ацетилхолина, снижение уровня которого в условиях инсулинорезистентности может усугублять течение БА.
Нарушение работы сигнальных систем инсулина сопровождается окислительным стрессом и митохондриальной дисфункцией, что дополнительно приводит к повышению экспрессии гена APP и продукции β-амилоида, замыкая еще один «порочный круг».
Наряду с ингибированием IRS-1 другим механизмом развития инсулинорезистентности при БА является перемещение инсулинового рецептора с клеточной мембраны внутрь клетки в результате его связывания с β-амилоидными олигомерами, что подтверждается данными аутопсии. Кроме того, растворимые олигомеры β-амилоида могут связываться с NMDA- и AMPA-рецепторами, индуцировать процесс эксайтотоксичности, а также повреждение постсинаптической мембраны с запуском механизмов апоптоза.
Настоящее и будущее: перспективы терапии
В настоящий момент ведутся активные поиски терапевтических стратегий, направленных на различные этапы патогенетического каскада, с тем чтобы отсрочить или остановить развитие деменции, которая наступает как минимум через 20 лет от момента запуска повреждающего механизма. Получены первые положительные результаты антиамилоидной терапии, основанной на пассивной и активной иммунизации, однако в целом эффективность предложенных средств оказалась недостаточной, а нежелательные явления неприемлемыми. Перспективы более ранней диагностики БА, основанной на применении нейровизуализационных и лабораторных биомаркеров, позволяют надеяться на более успешные результаты антиамилоидной терапии, однако перспектива выхода этих препаратов на рынок пока видится туманной.
Противодействовать избыточной аккумуляции и агрегации β-амилоида можно также за счет усиления его клиренса. Так, на этапе клинических испытаний находится ингибитор рецепторов конечных продуктов гликирования, который может влиять на выведение β-амилоида методом трансцитоза. Перспективны клинические испытания ингибитора АРР-трансформирующего фермента (BACE), который блокирует избыточное образование β-амилоида из АРР. Исследуется возможность усиления деградации агрегатов, например с помощью фермента неприлизина.
Таким образом, современная практика терапии БА продолжает основываться на применении 2 групп препаратов с преимущественно симптоматическим эффектом - ингибиторов холинэстеразы и модулятора глутаматных репторов NMDA-типа (мемантин), эффективность которых доказана в клинических плацебо-контролируемых испытаниях. Основной механизм их действия направлен на коррекцию нейромедиаторного дефицита, что позволяет замедлять скорость когнитивного снижения и улучшать показатели нейропсихологических шкал и повседневной активности пациентов. К сожалению, эффект обеих групп препаратов как в форме монотерапии, так и при совместном применении как минимум у трети больных не достигает клинической значимости, что оправдывает практическое применение при БА препаратов с иным механизмом действия.
Учитывая сложность, каскадность и многофакторность патогенеза БА, отражающих современное состояние амилоидной гипотезы, перспективным представляется применение препаратов с мультимодальным действием, среди которых особое внимание привлекает актовегин - депротеинизированный ультрафильтрат, получаемый из крови телят и состоящий из более чем 200 биологически активных компонентов.
Актовегин увеличивает утилизацию клетками кислорода, благодаря чему активирует энергетический метаболизм, переводя энергообмен клеток в сторону аэробного гликолиза, снижая образование лактата и тормозя окисление свободных жирных кислот. В результате, в условиях ишемии увеличивается содержание высокоэнергетичных фосфатов (АТФ и АДФ), что восполняет энергетический дефицит [32, 33]. Учитывая роль инсулинорезистентности в развитии как сосудистого, так и дегенеративного поражений мозга, важным представляется инсулиноподобное действие препарата, стимулирующего транспорт глюкозы через мембрану посредством активации белков-переносчиков GLUT-1,4, не задействуя при этом рецепторы инсулина [34]. В эксперименте с индуцированием гибели нейронов гиппокампа амилоидным белком Aβ25−35 было показано, что актовегин способствует сохранению жизнеспособности нейронов, за счет снижения уровня активированной каспазы-3, проявляя тем самым нейропротективные свойства [35]. Помимо этого недавно установлено, что актовегин ингибирует активность полимеразы поли-АДФ-рибозы - ядерного фермента, избыточная активация которого может запускать процессы клеточной гибели при ишемии мозга. На модели in vitro показана способность препарата актовегин модулировать активность нуклеарного фактора NF-kB, играющего важную роль в регуляции процессов апоптоза и воспаления [36, 37].
С прагматической точки зрения, особую ценность актовегин приобретает в связи с тем, что имеет доказательства клинической эффективности, полученные в плацебо-контролируемых испытаниях при различных нозологических формах деменций, что позволяет использовать его в РФ в рамках их симптоматической терапии уже в настоящий момент. Так, по результатам систематического обзора, актовегин может быть полезен в лечении легкой и умеренной деменции без разделения на нозологические формы [38]. По результатам недавно завершившегося многоцентрового рандомизированного исследования ARTEMIDA, в котором изучалась эффективность препарата актовегин у пациентов с постинсультными когнитивными нарушениями при назначении в остром периоде инсульта, на фоне введения препарата отмечено улучшение состояния когнитивных функций по сравнению с плацебо спустя 6 мес терапии, а также наблюдалась тенденция к снижению числа пациентов с диагнозом деменция через 6 мес последующего наблюдения за данными пациентами [39].
Смерть после жизни, болезнь Альцгеймера и почему мы хотим перемен

Обзор
Автор
Редактор
Статья на конкурс «био/мол/текст»: Возможно, всякий мир чем-то сродни слепцу, попавшему в гигантскую паутину. Чем дальше мир двигается, тем больше запутывается в эластичных, клейких ниточках пространства-времени. Эти ниточки влияют на происходящие события, иногда они растягиваются, иногда рвутся или переплетаются, создавая новые формы и образы. А может, мы всё переусложняем. Известный философ Дидактилос выразил альтернативную гипотезу следующей ёмкой фразой: «Да какого чёрта, события просто случаются!»
Болезнь Альцгеймера, загадочная и опасная. Мы не знаем ее причин и лишь предлагаем гипотезы. Переусложняем ли мы всё или слишком упрощаем для удобства работы? Почему бета-амилоидная гипотеза должна уйти, узнаем в этом обзоре.
Обратите внимание!
Эта работа опубликована в номинации «лучшая обзорная статья» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Перед тем, как человек умрет, вся жизнь
действительно проносится у него перед глазами.
Собственно, этот процесс и называется жизнью.
Терри Пратчетт. Последний континент
(Introitus)
Этот рассказ — о памяти. И кое-что можно сразу припомнить.
«"Но могут ли существовать черепаха в десять тысяч миль длиной и слоны высотой более чем в две тысячи миль?" — удивляются люди. Это их удивление еще раз доказывает, что человеческий мозг плохо приспособлен для мыслительных процессов — скорее всего, изначально он был предназначен для охлаждения проходящей через него крови. Первым делом человеческий мозг изумляют размеры. Тогда как в размерах нет ничего удивительного. Куда больше поражают сами черепахи, да и слоны — крайне замечательные звери. А тот факт, что где-то во вселенной живет очень большая черепаха, куда менее примечателен, чем факт существования самой обычной черепашки» [1].
А наш разговор, конечно, пойдет о мозге, уютно покоящемся в черепной коробке. В этой озаренной электрическими импульсами комнате хранятся «жизнеизмерители». Клетка за клеткой, они сплетаются в запутанные сети, формируют строгие колонки и рисуют сложнейшие карты. Эти клетки — нейроны, — по сути, повторяют жизненный путь всего человека: они рождаются, несколько раз делятся еще во время эмбрионального развития (хотя из любого правила есть исключения), а дальше «остаются в нас до исполненья [мечты] или до смерти — это всё равно», жертвуя возможностью давать потомков ради создания прочной и надежной сети связей. И пускай наш мозг надежно защищен стеной гематоэнцефалического барьера, укрыт прочными костями черепа и избалован относительно бережным отношением к себе «организма-носителя», чего зачастую не хватает другим органам, нейроны неизбежно накапливают повреждения (в частности, в структуре митохондрий) и погибают достойной смертью на своем посту.
Старение человека всегда сопровождается медленной гибелью нейронов. Но зачастую они могут гибнуть и быстрее — целыми звездными скоплениями уходя в черную пустыню, под покров бескрайней ночи, оставляя разрывы и мертвые участки в некогда отлично работавшей нейронной сети. Подобный процесс ведет к тяжелым повреждениям памяти и нарушениям поведения. Такие ситуации называют нейродегенеративными заболеваниями — от словосочетания «дегенерация нейронов». И если речь в статье заходит о гибели нейронов, почти наверняка хотя бы раз в ней упомянут болезнь Альцгеймера.
(Kyrie)
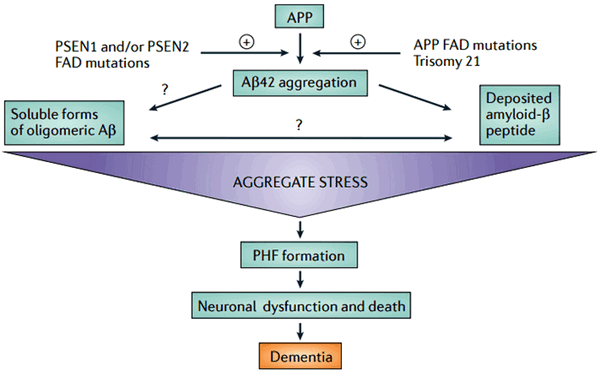
Рисунок 1. Общая схема развития заболевания согласно бета-амилоидной гипотезе. Белок — предшественник амилоида (АПП) разрезается по «амилоидогенному» пути с образованием молекул Aβ длиной 42 аминокислоты. Этот процесс может усиливаться при наличии мутаций, связанных с этим разрезанием или увеличением числа молекул АПП. Скопления амилоида в виде бляшек либо растворимых некрупных скоплений приводят к состоянию окислительного стресса, образованию фибрилл тау-белка и гибели нейронов. Обратите внимание на знаки вопроса. Рисунок из [33].
(Tract)
«Очень важно знать, откуда мы пришли, потому что если не знаешь, откуда ты, то не знаешь, где ты, а если не знаешь, где ты, то не знаешь и куда идёшь. А если ты не знаешь, куда идёшь, то, скорее всего, идёшь не туда» [7]. Поэтому давайте поподробнее рассмотрим все молекулярные симптомы, связанные с болезнью Альцгеймера, — с тем чтобы постараться определить возможную отправную точку в патогенезе болезни. Симптомы эти весьма разнообразны: помимо упомянутых скоплений Aβ и нарушений в работе холинергической системы, чаще всего упоминаются внутриклеточные фибриллы тау-белка и состояние окислительного стресса, в котором находятся нейроны при БА; реже, хотя и несправедливо, заходит речь о нарушениях в метаболизме ионов металлов (железа, цинка и меди), повреждении структуры митохондрий, накоплении липофусцина в клеточных везикулах и патологическом возвращении отдельных нейронов в клеточный цикл.
(Sequence)
Всё где-то начинается, хотя большинство ученых-физиков с этим не согласны. Вопрос о начале всегда бередил людские умы. «Вот, к примеру, — задаемся вопросом мы, — как водитель трактора, расчищающего снег, попадает на работу?» Или: «Откуда составители всевозможных словарей знают, что данное слово пишется именно так, а не иначе?» Нас преследует непреодолимое желание найти точку в переплетающихся, крайне запутанных нитях пространства-времени, в которую можно ткнуть метафорическим пальцем и воскликнуть: ага, именно здесь-то всё и началось [8]! Восклицание авторов бета-амилоидной гипотезы 1992 года было услышано, и следующие 20 лет ученые со всего мира тщательно изучали свойства Aβ. В ходе исследований обнаружилось, например, что примерно у 5% больных заболевание вызвано мутацией в одном из нескольких генов (APP, PS1 и PS2), тесно связанных с продукцией амилоида (это, кстати, не слишком характерно для исследований заболеваний человека: чаще вначале описывают моногенную форму болезни, которая и указывает на главный патологический фактор у всех больных). Весомое подтверждение гипотезы. Вдобавок, введение амилоида Aβ мышам и вправду вызывало у них когнитивные нарушения и проблемы со здоровьем, похожие на человеческую болезнь Альцгеймера. Более того, чрезмерная (овер-) экспрессия белка — предшественника амилоида (АПП) и у мышей, и даже у плодовых мушек дрозофил приводила к нейродегенерации (по понятным причинам — чем больше АПП, тем чаще он будет разрезаться с образованием бета-амилоида).
В норме трансмембранный белок АПП разрезается α-секретазой (обычно — ADAM10, цинк-содержащей металлопротеазой из семейства ADAM) с образованием растворимого фрагмента — sAPPα, обладающего нейротрофическими и нейропротекторными свойствами. С α-секретазой за субстрат (АПП) конкурирует β-секретаза (BACE1), образующая другую растворимую форму — sAPPβ, а совместно с γ-секретазным комплексом — и бета-амилоид (Aβ) [9]. К тому же недавно была описана еще одна протеаза — меприн-β, которая также разрезает АПП с образованием различных форм бета-амилоида [10]. Таким образом, образование Aβ — это процесс, протекающий в той или иной степени даже в норме (хотя в обычных условиях α-секретаза работает значительно эффективнее). А при изменении условий (например, при снижении pH внутри нейрона и появлении там больших концентраций свободных ионов цинка) равновесие резко сдвигается в сторону усиления активности β-секретазы и увеличения продукции Aβ [11].
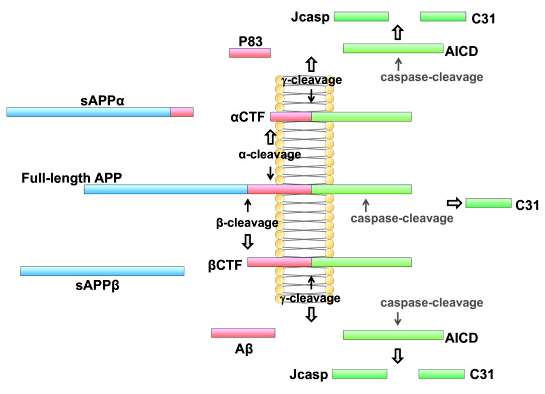
Рисунок 2. Общая схема процессинга молекулы АПП. При разрезании этого белка α-секретазой образуется нейропротекторный секреторный компонент α, sAPPα (считается, что это происходит на поверхности клетки). В результате работы β-секретазы образуется секреторный компонент β, sAPPβ (регулирующий экспрессию некоторых генов, проапоптотический), а после разрезания оставшегося от него мембранного фрагмента АПП γ-секретазой высвобождается бета-амилоид, Aβ (считается, что это происходит в основном в эндоплазматической сети и аппарате Гольджи). CTF — C-концевой фрагмент. Рисунок из [34].
Еще Aβ оказался токсичным для нейронов, амилоидные скопления гораздо чаще встречались у больных БА, а изоформа АпоЕ4 знаменитого белка АпоЕ (носительство аллели APOЕ4 говорит о повышенном риске заболеть БА) с транспортом амилоида из мозга справляется заметно хуже, чем ее «нормальная» сестрица — АпоЕ3 [12].
Казалось бы, всё здесь понятно. Однако всё больше авторов с подобным подходом не согласны. Разберемся, с чем это связано. Благо, как было верно подмечено в статье Карла Эррапа (Karl Herrup, [13], за ссылками на бόльшую часть фактов из ближайших трех абзацев можно обратиться к ней), бета-амилоидная гипотеза, как любая хорошая гипотеза, выдвигает четкие и проверяемые тезисы. Вкратце, гипотеза постулирует, что:
- введение амилоида здоровым людям вызовет у них развитие болезни Альцгеймера;
- очистка мозга больных от бета-амилоида развитие болезни если и не обратит, то остановит.
И введение амилоида мышам болезнь действительно вызывает; более того, если этот амилоид потом убрать, болезнь полностью отступает (что логично). Проблема заключается в том, что у здоровых пожилых людей скопления амилоида зачастую есть (Хор: «Бляшки предшествуют развитию симптомов»). А еще — в том, что если от амилоида избавиться (а методов осуществления этого накопилось уже достаточно, включая любопытный метод иммунизации с получением антител к Aβ [14]), то ни улучшения состояния больных, ни полной остановки болезни не происходит (тем самым бета-амилоидную гипотезу формально уже можно считать опровергнутой).
И это самое главное, суть гипотезы. «В начале было ничто, которое взорвалось» [15]. Интересное с точки зрения физики наблюдение, которое в биологии обычно к верным выводам не приводит. Многочисленные авторы во введении к своим работам мимоходом описывают молекулярные механизмы образования Aβ и приступают к разбору его участия в очередном взаимодействии. Главный же вопрос — каким образом без каких-либо причин и поводов АПП вдруг массово и резко начинает разрезаться «неправильно»? А точнее, зачем организму закладывать часовую бомбу в виде сразу нескольких механизмов продукции сугубо токсичного бета-амилоида? И при каких условиях они срабатывают? Забавно, но авторы иногда даже оговариваются, что причины этого действительно неясны, что, возможно, это вызвано окислительным стрессом, гипоксией, воспалением или чем-то еще. Это, несомненно, может быть, однако и автоматически означает, что амилоид ПЕРВОПРИЧИНОЙ болезни не является. Более того, и воспаление, и окислительный стресс, и гипоксия — состояния весьма разнообразные и индивидуальные. Болезнь Альцгеймера же проявляется у огромного количества людей (по сути, у каждого третьего американца в возрасте свыше 85 лет и у каждого девятого старше 65 лет [16]) абсолютно одинаково в виде целого набора патологий — то есть первопричина, скорее всего, должна быть одна. И лежит она выше, чем бета-амилоид. Более того, всё чаще обсуждается антиоксидантная функция амилоида [17]. Тогда активация β-секретазы при снижении pH (читать как «при окислительном стрессе») выглядит весьма логичной, как и неудачи попыток лечения болезни путем полного избавления от амилоида. Что интересно, даже в случаях, когда при терапии удавалось избавиться от амилоидных бляшек, все когнитивные нарушения и прочие характерные для БА симптомы сохранялись [13].
(Offertorium)
К чему это всё? А к тому, что от самых красивых гипотез нужно уметь отказываться, если они не работают. Это утверждение, раньше робко читаемое между строк или упоминаемое с постоянными оговорками («Мы ни в коем случае не отрицаем амилоидную гипотезу, но вот в этом случае, возможно, она объясняет не всё»), дошло до заголовков в Nature Neuroscience ("The case for rejecting the amyloid cascade hypothesis" [13]) и других журналах: "Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’" [18], "Moving away from amyloid beta to move on in Alzheimer research" [19]. Пока большинство исследований остаются направленными на поиск способа борьбы с амилоидом (который, возможно, вообще играет антиоксидантную или иную защитную роль и не так плох, как кажется), а в клинические испытания входят всё новые препараты, которые не трогают первопричину и (как, к сожалению, пока выходит) не сильно облегчают симптомы, мы практически стоим на месте.

Рисунок 3. Иллюстрация художника Марка Симонетти (Marc Simonetti) к книге Т. Пратчетта «Мелкие боги». Люди начинают с веры в Бога и заканчивают верой в систему. И даже самые всемогущие существа, Великие Боги, могут исчезнуть и сгинуть в забытье, когда в них никто больше не верит. Как и гипотезы.
(Agnus Dei)
Вспомним про скопления тау-белка внутри нейронов — второй самый заметный патологический признак. Если насчет физиологической функции бета-амилоида ведутся споры, то с тау-белком таких вопросов нет — он очень хороший и полезный [20]. Например, он стабилизирует микротрубочки, что особенно важно для нейронов с их длинными отростками [21]. Регулируются функции тау-белка любимым клеткой способом — фосфорилированием тех или иных участков его молекулы, которых у тау-белка около пятидесяти. Активно фосфорилируется он, например, при подготовке клетки к делению — что стόит запомнить. И при болезни Альцгеймера его молекулы также гиперфосфорилируются, отходят от клеточного скелета и слипаются друг с другом, приводя к разборке микротрубочек и нарушениям процессов клеточного транспорта. Появление скоплений тау-белка по времени и своему положению в мозге очень хорошо коррелирует с развитием болезни Альцгеймера [22]. Конечно же, была предложена тау-гипотеза развития болезни [23]. Но будем справедливы, к ней тоже есть вопросы. Например, существует целый спектр таупатий — болезней, связанных с гиперфосфорилированием тау-белка, — и для всех из них, кроме болезни Альцгеймера, не характерно появление амилоидных скоплений и других патологий. Кроме того, имеются многочисленные данные о том, что бета-амилоид тем или иным способом стимулирует фосфорилирование тау-белка [24, 25]. Данный факт, во-первых, подвергает сомнению первичную роль тау-белка в патогенезе, а во-вторых, указывает на важность амилоида для этого процесса.
Кстати, в нейронах, содержащих фибриллы тау-белка, наблюдается еще одна любопытная вещь — эти клетки содержат целый набор маркеров, характерных для готовящихся к делению клеток (напомню, что способность к делению — мягко говоря, не сильная сторона нейронов) [26]. Более того, в таких клетках даже происходит удвоение (репликация) ДНК, хотя потом, конечно, они всё же застревают перед самым митозом. Вполне вероятно, что амилоид стимулирует возвращение нейронов в клеточный цикл [27] — и этому может быть физиологическое объяснение. Но об этом в другой раз.
(Communio)
Что же мы получаем в итоге? Что бета-амилоид, конечно, один из ключевых участников развития болезни Альцгеймера — но участник (возможно невольный, убивающий в попытке защитить), вовлеченный в процесс на более поздних стадиях. Что же могло быть в начале? Например, повреждение структуры митохондрий и накопление мутаций в их ДНК [28, 29], что происходит и при «нормальном» старении (а болезнь Альцгеймера, напомню, четко ассоциирована с возрастом, в большинстве случаев она проявляется после 65 лет). Подобное нарушение ведет к усиленной продукции токсичных для клетки радикалов и возникновению условий окислительного стресса. При этом, несмотря на то, что мутации в мтДНК появляются случайно, практически все они ведут к нарушениям в работе электрон-транспортной цепи митохондрий. Результат этого всегда одинаков: усиление продукции токсичных радикалов и снижение — АТФ. Это неплохо объясняет однообразие набора патологий у многочисленных больных. Митохондриальная гипотеза болезни Альцгеймера подробно разобрана в статьях Рассела Свердлова [30]. Образование амилоида в этом случае можно было бы объяснить чрезмерной активацией защитных механизмов — и хороший, полезный антиоксидант-амилоид, по аналогии с витамином Е, в избыточных концентрациях становится мощным окислителем. Однако в любом случае всегда нужно помнить, что нельзя останавливаться на одной гипотезе. Бета-амилоидная гипотеза дала стимул многим крайне важным для науки и для нашего понимания болезни Альцгеймера исследованиям*. Теперь, пожалуй, настало время идти дальше.
* — И понимания других загадочных недугов тоже. Детальное изучение «родственных связей» неинфекционных амилоидов и их инфекционных коллег — прионов — проведено автором статьи «Прионные и неприонные амилоиды: определяет ли конформация разницу в инфекционности?» [31]. — Ред.
(In paradisum)
Эту статью я пишу в память о сэре Терри Пратчетте, который скончался от болезни Альцгеймера 12 марта 2015 года. «Терри взял Смерть за руку и прошел с ним через двери в черную пустыню под покровом бескрайней ночи» [32]. Конец.
β-амилоид: невидимый враг или тайный защитник? Запутанная тропка болезни Альцгеймера

Новость
Авторы
Редакторы
Болезнь Альцгемера — нейродегенеративное заболевание с крайне удручающими симптомами: пациенты становятся беспомощными, утрачивают связь с реальностью и даже теряют способность говорить. В 2016 году научным коллективам удалось немного продвинуться как в понимании молекулярных основ этой патологии, так и в разработке способов борьбы с ней. В частности, были проведены клинические испытания сразу двух препаратов, направленных на уничтожение β-амилоидных частиц — структур, связанных с развитием болезни Альцгеймера. В то же время эксперименты на живых организмах подтвердили предположение о том, что β-амилоид обладает полезными для нашего организма свойствами — он является важным элементом врожденного иммунитета.

12 биологических новостей в картинках
Вообще, мы серьезные люди. Гранит науки хрустит на наших зубах. Мы освещаем такие суровые, такие сложные закоулки биологического знания, до которых не дотянулись фонари других научно-популярных сайтов. Но иногда нам так хочется подурачиться. И рассказать о науке веселым языком, показать ее под другим углом. Нарисовать забавных картинок, написать легкий и смешной текст. Поэтому мы и открыли новую рубрику — «12 биологических новостей в картинках».
Интеллектуальный партнер этих иллюстрированных рассказов — АО РВК.
Болезнь Альцгеймера, как правило, развивается у людей старше 65 лет. Внешне она проявляется триадой «афазия-апраксия-агнозия»: человек утрачивает способность совершать элементарные действия, распознавать предметы и лица людей, нарушается его речевая активность. При этом отклонениям в поведении пациентов предшествуют физиологические изменения в их организме. За 10-15 лет до первых проявлений заболевания в мозге начинают откладываться так называемые β-амилоидные бляшки. Эти характерные скопления сформированы преимущественно β-амилоидом — пептидом, образующимся особым ферментативным расщеплением белка — предшественника β-амилоида. Многие исследователи считают, что эти образования способствуют прогрессирующей дегенерации клеток мозга, в связи с чем разрабатывают лекарства, направленные на уничтожение или уменьшение числа β-амилоидных скоплений. В 2016 году сразу два таких противоамилоидных препарата проходили клинические испытания.

Чтобы увидеть рисунок в полном размере, нажмите на него.
Одно из этих лекарств разработали в Швейцарии. Вначале ученые отбирали образцы крови у пожилых людей, которые не демонстрировали никаких когнитивных нарушений. Затем из отобранной крови они выделяли B-лимфоциты. При этом важно было выделить такие клетки, антитела которых распознавали бы «токсичные» β-амилоидные бляшки, но не белок-предшественник. Дело в том, что предшественник β-амилоида присутствует во всем организме и играет важную роль в росте нервных клеток, поэтому затрагивать его крайне нежелательно — можно навредить здоровью. В результате было получено моноклональное антитело адуканумаб (aducanumab) [1]. Оно избирательно реагирует с вредным β-амилоидом, но не атакует полезный белок-предшественник. Предположительно, это антитело может помочь пациентам на ранней стадии болезни Альцгеймера.
К настоящему моменту уже завершена первая фаза клинических испытаний адуканумаба [2], [3]. Лечению этим антителом подвергались 165 пациентов на ранней стадии болезни Альцгеймера: часть из них получала адуканумаб, а часть (контрольная группа) — плацебо. Сравнивая между собой такие группы, можно судить об эффективности лекарства. Оказалось, что у пациентов из группы плацебо значительно снижались когнитивные способности, тогда как у пациентов, получающих антитело, они стабилизировались. Проверяли это при помощи стандартных вопросников для оценки познавательных способностей. Также методом позитронно-эмиссионной томографии (ПЭТ) ученые выяснили, что адуканумаб способствует уменьшению β-амилоидных отложений в мозге. В настоящее время проводятся новые клинические исследования для дальнейшей оценки безопасности и эффективности адуканумаба.

Второй препарат получили исследователи из США и Канады [4]. Называется он верубецестат (verubecestat), и его функция заключается в ингибировании фермента бета-секретазы (BACE1). Этот фермент — один из главных «производителей» β-амилоида в мозге. Исследования, которые проводились на грызунах и приматах, показали, что у животных, принимавших верубецестат на протяжении 6-9 месяцев, содержание β-амилоида в центральной нервной системе заметно снижалось. К сожалению, результаты оказались не столь вдохновляющими, когда очередь дошла до лечения людей. В феврале 2017 года было решено прекратить клинические испытания верубецестата, поскольку препарат не проявлял эффективности в борьбе с уже развившейся болезнью Альцгеймера — по крайней мере, в случае ранней или умеренной деменции. Независимые эксперты сошлись во мнении, что в данном случае шансов получить положительный клинический эффект практически нет. Однако исследователи не теряют надежды: есть вероятность, что верубецестат подойдет для лечения пациентов с первыми проявлениями болезни Альцгеймера. Результаты этих клинических испытаний будут известны в феврале 2019 года.
В последнее время амилоидная гипотеза развития болезни Альцгеймера постепенно теряет свои позиции. Один из главных фактов, опровергающих устойчивое мнение о вредоносности β-амилоида, обнаружили исследователи из Массачусетской клинической больницы [5], [6]. Оказалось, что β-амилоид представляет собой нормальный компонент врожденного иммунитета. Ранее ученые подтвердили это предположение в экспериментах in vitro, то есть продемонстрировали соответствующие эффекты в модельных системах вне живого организма [7]. Сейчас же они получили аналогичные результаты в опытах in vivo, то есть на живых организмах. Новые исследования на мышах и круглых червях, как и эксперименты с культивируемыми клетками человеческого мозга, показали, что β-амилоид способен защищать организм от потенциально летальных инфекций. Ученые сравнивали синтетические формы β-амилоида с кателицидином — антимикробным пептидом, который помогает справляться с инвазивными бактериальными инфекциями. Оказалось, что β-амилоид ингибирует рост нескольких опасных патогенов так же, а иногда даже и лучше, чем кателицидин.
β-Амилоид, выделенный из мозга пациентов с болезнью Альцгеймера, подавлял рост культивируемых дрожжей Candida, а его синтетический аналог оказался эффективен против вирусов гриппа и герпеса. Также ученые выяснили, что трансгенные мыши, у которых экспрессировался ген человеческого β-амилоида, жили после заражения сальмонеллой значительно дольше их обычных сородичей. А быстрее всех погибали мыши, лишенные какого бы то ни было амилоидного предшественника. Трансгенная экспрессия β-амилоида защищала круглых червей и культуры нейронов от заражения микроорганизмами Candida и Salmonella. И, как вишенка на торте, последний факт: человеческий β-амилоид, синтезируемый живыми клетками, оказался в 1000 раз эффективнее в борьбе с инфекциями по сравнению с его синтетическими аналогами.
Почему так происходит? Вероятно, антимикробный эффект β-амилоида объясняется как раз его способностью образовывать агрегаты: его молекулы связываются с поверхностью микробов, а затем объединяются в плотные структуры, что предотвращает прикрепление патогенных микроорганизмов к клеткам макроорганизма. Тогда почему же амилоидные бляшки накапливаются именно в мозге? Ученые не исключают следующий вариант: возможно, мозг по каким-то причинам начинает воспринимать себя «атакованным» вторгающимися патогенами (и, возможно, в действительности так оно и есть), в результате чего запускается усиленная выработка амилоида. Но для подтверждения этой гипотезы требуются дальнейшие исследования.
Так каков же итог? Как лечить болезнь Альцгеймера? Пока очевидного ответа на этот вопрос, увы, нет. Однако, учитывая новые данные, следует понимать: тотальное уничтожение β-амилоида может быть чревато негативными последствиями для здоровья. Амилоидная гипотеза выглядит уже не так многообещающе, и ученым предстоит искать новые мишени для лечения этого тяжелого недуга. Хочется верить, что эра новых лекарств против болезни Альцгеймера уже не за горами.
Читайте также: