Болезнь Кавасаки (острый фебрильный слизисто-кожный синдром): клиника, диагностика
Добавил пользователь Alex Обновлено: 21.01.2026
Слизисто-кожный лимфонодулярный синдром (синдром/болезнь Кавасаки) представляет собой остро протекающее системное заболевание, характеризующееся преимущественным поражением средних и мелких артерий (артериит), развитием деструктивно-пролиферативного васкулита. Иногда в процесс могут вовлекаться аорта и другие крупные артерии. Наиболее часто синдром Кавасаки встречается у детей грудного и раннего возраста.
Синдром Кавасаки (СК) - один из диагнозов, который должен обязательно рассматриваться в качестве причины фебрильной лихорадки у детей. СК у детей, являясь относительно редкой патологией, может вызывать развитие аневризм и стенозов коронарных артерий, особенно при поздней диагностике и несвоевременном и/или неадекватном лечении. Таким образом, СК - одна из причин приобретенных сердечно-сосудистых заболеваний.
Изменения коронарных артерий, представляющие собой фактор риска летального исхода и инфаркта миокарда в молодом возрасте, у подавляющего большинства больных можно предупредить при условии своевременного (до 10 дня заболевания) лечения большими дозами внутривенного иммуноглобулина человека (ВВИГ) в комбинации с ацетилсалициловой кислотой.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
• Болезнь Кавасаки, полная форма от 11.2014 года. Синдром дилатационной кардиомиопатии. Аневризмы левой и правой коронарной артерий. Хроническая сердечная недостаточность (ХСН) IIа, функциональный класс (ФК) II по Ross.
• Болезнь Кавасаки, неполная форма от 05.2013 года. Окклюзия правой коронарной артерии. ХСН I, ФК I по NYHA.
Европейским обществом детских ревматологов (Paediatric Rheumatology European Society - PReS) и Европейской лигой по проблемам ревматизма (European League against Rheumatism - EULAR) в 2006 г. принята следующая классификация васкулитов у детей: [13].
• Гипокомплементемический уртикарный васкулит
• Вторичные васкулиты при инфекциях (в том числе узелковый полиартериит, ассоциированный с гепатитом В), опухолях и лекарственные, включая васкулит гиперчувствительности
Сердечно-сосудистые нарушения, возникающие вследствие СК, классифицируют в соответствии с размерами аневризм и тяжестью проявлений сердечно-сосудистых нарушений [14]
У детей ≥5 лет: внутренний диаметр измеряемого сегмента отличается от внутреннего диаметра примыкающего сегмента менее, чем в 1,5 раза.
У детей ≥5 лет: внутренний диаметр измеряемого сегмента отличается от внутреннего диаметра примыкающего сегмента в 1,5-4 раза
У детей ≥5 лет: внутренний диаметр измеряемого сегмента отличается от внутреннего диаметра примыкающего сегмента более, чем в 4 раза
B. В соответствии со степенью тяжести проявлений сердечно-сосудистых нарушений при СК выделяют группы от I до V.
Определение группы тяжести - на основании данных электрокардиографии (Эхо-КГ) и селективной ангиографии или других методов:
I. Нет дилатаций коронарных артерий: пациенты без дилатаций коронарных артерий, в том числе, в острую фазу болезни;
II. Транзиторная дилатация коронарных артерий в острую фазу болезни: пациенты со слабовыраженными и транзиторными дилатациями, исчезающими, как правило, в течение 30 дней от их появления;
III. Регрессия: пациенты, у которых еще определяются аневризмы коронарных артерий, соответствующие критериям дилатации или более выраженные изменения на 30 день после их появления, несмотря на полное исчезновение изменений в билатеральных системах коронарных артерий в течение первого года после их появления, а также пациенты, у которых изменения коронарных артерий не удовлетворяют критериям для включения в группу V;
IV. Сохраняющиеся аневризмы коронарных артерий: пациенты у которых определяются одно-или двусторонние аневризмы коронарных артерий по данным коронарной ангиографии на втором году после острой фазы СК или позже и пациенты, у которых изменения коронарных артерий не удовлетворяют критериям для включения в группу V;
V. Стеноз коронарных артерий: пациенты со стенозом коронарных артерий, подтвержденными коронарной ангиографией:
(a) Пациенты без признаков/симптомов ишемии, подтвержденной лабораторными тестами или другими методами;
(b) Пациенты с признаками/симптомами ишемии, подтвержденными лабораторными тестами или другими методами;
• если у пациентов имеются среднетяжелые или тяжелые поражения клапанов сердца, сердечная недостаточность, тяжелая аритмия или другие сердечно-сосудистые заболевания, эти состояния должны быть учтены при оценке тяжести СК.
Этиология и патогенез
Этиология синдрома Кавасаки до настоящего времени окончательно не установлена. Авторы большинства многочисленных эпидемиологических и иммунологических исследований склоняются к тому, что наиболее вероятным причинным фактором может служить инфекционный агент (предположительно вирус) [1]. Кроме того, немаловажными факторами в развитии синдрома Кавасаки могут быть аутоиммунные механизмы и генетическая предрасположенность [2,3,4]. На сегодняшний момент имеются данные о 6 генетических локусах, связанных с этим заболеванием [5].
Эпидемиология
СК описан впервые Т.Kawasaki в 1967 г. в Японии; там, как и в странах Азии, данная патология встречается наиболее часто, что указывает на наличие генетической предрасположенности. В Японии заболеваемость составляет 137,7 на 100 тыс. детского населения по данным на 2002 г. и 218,6 - в 2008г [6], в США - 9 - 19 [7], на Тайване - 69 [8], в Великобритании - 8 на 100 тыс. детского населения [9].
Примерно 90-95% заболевших - дети в возрасте до 10 лет, до 85-90% случаев приходится на пациентов младше 5 лет. Наиболее часто болеют младенцы 9-11 мес.
В России СК диагностируется все чаще, однако нередко в поздние сроки, вследствие чего лечение назначается несвоевременно и не всегда адекватно [10,11]. По данным эпидемиологического исследования, проведенного в Иркутской области с 2005 по 2009г, средний уровень заболеваемости составил 2,7 на 100 тыс. детей от 0-17 лет и 6,6 среди детей младше5 лет, при этом авторы признают, что эти цифры могут быть заниженными [12].
Клиническая картина
Cимптомы, течение
Наиболее характерные проявления синдрома Кавасаки представлены в табл. 1.
Таблица 1 - Стадии синдрома Кавасаки [15]
В подостром периоде уже можно наблюдать расширение артерий - аневризмы, тромбозы, стеноз артерий среднего размера, панваскулит и отек сосудистой стенки; миокардит менее очевиден.
В дальнейшем воспалительные явления в сосудах уменьшаются, небольшие расширения подвергаются обратному развитию, но часть аневризм остается, угрожая тромбозом и инфарктом миокарда.
У 2,2% пациентов с СК обнаруженные с помощью ангиографии аневризмы не только в коронарных артериях, но и в подключичной, подмышечных, внутренней грудной артерии, почечной артерии, верхней брыжеечной артерии, общей подвздошной артерии, внутренней подвздошной артерии, бедренной артериях, имели гигантские размеры и множественный характер [19]. Аневризматическое расширение периферических сосудов иногда удается пальпировать.
Одна из проблем диагностики заключается в том, что проявления СК возникают последовательно, вследствие чего ранние из них, например, сыпь, могут быть не зафиксирована врачом. А наиболее часто обнаруживаемый признак - шелушение кожи на ладонях и стопах - выявляется в более поздней, подострой стадии, когда уже могут иметь место осложнения со стороны сердца.
Помимо классической формы, СК может протекать как «неполная форма», чаще у детей первых месяцев жизни [20]. По наблюдениям Национального научного-практического центра здоровья детей, атипичный СК отмечается у 20% больных [10]. Обычно кроме лихорадки имеют место не 4, а всего 2-3 признака: например, склерит и гиперемия кожи с припухлостью над межфаланговыми суставами кистей. Диагноз СК в этих случаях представляет трудности, иногда он становится очевидным при появлении дополнительных симптомов, в других постановке диагноза помогает исключение других причин стойкой лихорадки. В отдельных случаях при неполной клинической картине СК при ЭхоКГ выявлялись изменения стенок и диаметра коронарных артерий, что делало диагноз СК весьма вероятным [21]. Эти изменения и позже развивающиеся аневризмы коронарных артерий (АКА) - почти патогномоничный признак СК, поскольку аневризмы артерий, не связанные с СК, встречаются у детей нечасто (в аорте при ее коарктации, внутричерепных сосудах при синдроме Марфана, а также при бактериальных эмболах артерий, узелковом полиартериите или аортоартериите, имеющих иную клиническую картину).
Необычно начало СК с картины заглоточного абсцесса (лихорадка, болезненность при поворотах головы, тризм) с гипоэхогенным (нативная КТ плотность 20-30 ед) линзообразным, не накапливающим контраст скоплением в заглоточной области на КТ. Отсутствие гноя при вскрытии припухлости на задней стенке глотки и сохранение температуры несмотря на антибактериальную терапию, легкий склерит и эффект от введения 2 г/кг ВВИГ, а также шелушение кожи ладонной поверхности пальцев позволяют подтвердить диагноз СК [22].
Диагностика
1. Лихорадка, часто до 40 С˚ и выше, длительностью минимум 5 дней и наличие хотя бы четырех из приведенных ниже пяти признаков:
2. Изменения слизистых оболочек, особенно ротовой полости и дыхательных путей, сухие, в трещинах губы; «земляничный» /малиновый язык, гиперемия губ и ротоглотки.
3. Изменения кожи кистей, стоп, (в том числе плотный отек, покраснение ладоней и подошв, часто - яркая эритема над мелкими суставами кистей и стоп) в ранней фазе, а также генерализованное или локализованное шелушение в паховых областях и на подушечках пальцев рук и ног на 14-21-й день от начала заболевания.
4. Изменение со стороны глаз, прежде всего двусторонняя инъекция сосудов склер и конъюнктивы, без слезотечения и изъязвления роговицы; при осмотре в проходящем свете может быть выявлен передний увеит.
5. Увеличение размеров лимфоузлов (в 50% случаев), особенно шейных, чаще возникает одиночный болезненный узел диаметром более 1,5 см.
6. Сыпь, которая появляется в первые несколько дней болезни и угасает через неделю; сыпь чаще диффузная, полиморфная - макулопапулезная, уртикарная, скарлатиноподобная или даже кореподобная без везикул или корочек.
Кроме вышеуказанных симптомов, рекомендуется также при наличии следующих симптомов и признаков рассмотреть вероятность синдрома Кавасаки у ребенка [24]:
1. Сердечно-сосудистая система: аускультация (сердечный шум, ритм галопа,), изменения на ЭКГ (удлинение интервалов PR/QT аномальная Q волна, низкий вольтаж комплекса QRS, изменения сегмента ST и T-зубца, аритмии), кардиомегалия по данным обзорной рентгенограммы органов грудной клетки, ЭхоКГ (жидкость в полости перикарда, аневризмы коронарных сосудов), аневризмы периферических артерий (например, аксиллярной), загрудинные боли (стенокардия) или инфаркт миокарда
2. Желудочно-кишечный тракт: диарея, рвота, боль в животе, водянка желчного пузыря, паралитический илеус, легкая желтушность кожи, небольшое кратковременное повышение сывороточных трансаминаз.
3. Кровь: лейкоцитоз со сдвигом влево, тромбоцитоз (до 1-1,2 млн), ускорение СОЭ, повышение уровня СРБ, гипоальбуминемия, повышение уровня α2-глобулина, небольшое повышение количества эритроцитов и уровня гемоглобина
5. Кожа: гиперемия и появление корки на месте введения БЦЖ вакцины мелкие пустулы, поперечные борозды на ногтях пальцев рук.
6. Органы дыхания: кашель, ринорея, затемнения легочных полей на обзорной ренгенограмме органов грудной клетки
8. Неврологические: плеоцитоз в цереброспинальной жидкости (с преобладанием мононуклеаров с нормальным уровнем белка и углеводов), судороги, потеря сознания, паралич лицевого нерва, паралич конечностей
Следует подчеркнуть важность выявления склерита для предположения о СК при скудности или необычности другой симптоматики. Обнаружение при ультразвуковом исследовании расширения или, хотя бы, изменений стенок коронарных артерий, позволяет подтвердить диагноз СК при наличии лишь 2 признаков из 6.
Самый существенный признак СК - стойкая лихорадка, которая начинается, как правило, внезапно, достигая 40˚С и выше, резистентная к жаропонижающим препаратам. Ее «диагностический минимум» - 5 дней, но обычно она держится намного дольше, иногда на протяжении месяца. На фоне лихорадки в течение первых 10 дней обычно появляются симптомы, относящиеся к основным критериям диагностики заболевания (типичные признаки СК): сыпь, сухие в трещинах гиперемированные губы, гиперемия и инъецированность склер, плотный отек и покраснение ладоней и подошв.
Характерный клинический признак для детей раннего возраста - покраснение и уплотнение места инъекции БЦЖ (этот признак не был внесен в список обязательных, т.к. в США нет массовой вакцинации БЦЖ).
Следует тщательно провести расспрос родителей (законных представителей) с целью выявления анамнестических данных о типичных и/или вероятных проявлениях СК.
Необходимо проведение стандартного осмотра ребенка. Обязательно обратить внимание на типичные признаки СК.
Клинически может выявляться тахикардия, аритмия (вследствие вовлечения в процесс проводящей системы сердца, вплоть до развития угрожающих жизни аритмий), выслушиваться шумы в сердце вследствие поражения клапанного аппарата (митральная, аортальная, трикуспидальная недостаточность, как правило, обратимые без формирования клапанных пороков), возможно развитие сердечной недостаточности.
Одна из проблем диагностики заключается в том, что проявления СК возникают последовательно, вследствие чего, ранние проявления, например, сыпь, может быть не зафиксирована врачом. А наиболее часто обнаруживаемый признак - шелушение кожи на ладонях и стопах, выявляется в более поздней, подострой стадии, когда уже могут иметь место осложнения со стороны сердца.
• Рекомендовано проведение следующих лабораторных тестов (Американская Ассоциация Кардиологов (American Heart Association - AHA) и Американская Академия Педиатрии (American Academy of Pediatrics - AAP)), особенно у пациентов с вероятным неполным синдромом Кавасаки [23,25]:
Болезнь Кавасаки
Болезнь Кавасаки — редкое иммунокомплексное воспалительное поражение артерий различного калибра, возникающее преимущественно у детей первых лет жизни. Болезнь Кавасаки проявляется лихорадкой, полиморфной диффузной сыпью, конъюнктивитом, поражением слизистой рта, кожи и суставов дистальных отделов конечностей, шейной аденопатией. Диагностика болезни Кавасаки основана на клинических критериях, результатах лабораторных исследований крови и мочи, данных ЭКГ, УЗИ сердца и коронарографии. Основу лечения болезни Кавасаки составляет внутривенное введение иммуноглобулина и прием ацетилсалициловой кислоты, по показаниям применяются антикоагулянты.
Общие сведения
Болезнь Кавасаки получила свое название благодаря открывшему ее в 1961 году японскому педиатру по фамилии Кавасаки. Первоначально предполагалось, что заболевание имеет легкое течение. Лишь в 1965 году был выявлен случай тяжелой сердечной патологии, связанный с перенесенной болезнью Кавасаки. В России первый клинический случай болезни Кавасаки был диагностирован в 1980 году.
Сегодня болезнь Кавасаки является одной из самых распространенных причин приобретенной патологии сердца в детском возрасте. Наиболее часто заболевание встречается среди представителей желтой расы, особенно японцев. В Японии болезнь Кавасаки диагностируется в 30 раз чаще, чем в Австралии или Великобритании и в 10 раз чаще, чем в Америке.
Причины возникновения болезни Кавасаки
Симптомы болезни Кавасаки
Болезнь Кавасаки начинается с подъема температуры тела. Без лечения лихорадка сохраняется в течение 2-х недель. Увеличение лихорадочного периода считается прогностически неблагоприятным симптомом.
Кожные проявления болезни Кавасаки могут возникнуть в течение 5 недель от начала заболевания. Они характеризуются полиморфными диффузными элементами в виде мелких плоских пятен красного цвета (макулярная сыпь), волдырей, скарлатиноподобных или похожих на корь высыпаний. Элементы сыпи располагаются в основном на коже туловища, паховой области и проксимальных отделов конечностей. Со временем возникают эритематозные участки, отмечается болезненное уплотнение кожи ладоней и подошв, обуславливающее ограничение движений в пальцах. Разрешение элементов сыпи при болезни Кавасаки начинается примерно через неделю после их появления. Эритематозные пятна сохраняются 2-3 недели, после чего их поверхность начинает шелушиться.
Поражения слизистых оболочек глаз и ротовой полости. У большинства заболевших болезнью Кавасаки в течение первых 7 дней отмечается появление конъюнктивита обоих глаз, обычно не сопровождающегося выделениями. В некоторых случаях ему сопутствует передний увеит. Наблюдается также сухость и покраснение слизистой оболочки ротовой полости, кровоточащие трещины на губах, малиновая окраска языка и увеличение миндалин. Болезнь Кавасаки в 50% случаев сопровождается увеличением шейных групп лимфатических узлов, чаще односторонним.
Поражение сердца и сосудов при болезни Кавасаки может носить характер миокардита, проявляющегося тахикардией, болями в сердце, аритмией и часто приводящего к острой сердечной недостаточности. У 25% пациентов с болезнью Кавасаки через 5-7 недель от начала заболевания выявляются аневризматические расширения коронарных сосудов сердца, которые могут приводить к развитию инфаркта миокарда. В редких случаях появляется перикардит, аортальная или митральная недостаточность. Возможно возникновение аневризм по ходу крупных артерий: локтевых, подключичных, бедренных.
Суставной синдром наблюдается в 35% случаев болезни Кавасаки и длится обычно до 1 месяца. Типичны артралгии и артриты голеностопных и коленных суставов, поражения мелких суставов кистей и стоп.
Возможно поражение органов ЖКТ с возникновением болей в животе, рвоты, поноса. В отдельных случаях наблюдается менингит, уретрит.
Диагностика болезни Кавасаки
Общепринятыми клиническими диагностическими критериями болезни Кавасаки является наличие на фоне продолжающейся более 5 дней лихорадки как минимум 4 из ниже приведенных признаков.
- Двусторонний конъюнктивит.
- Полиморфная сыпь с диффузным распространением по кожному покрову.
- Поражение слизистой рта.
- Изменения кистей и стоп с их покраснением и отеком.
- Увеличение шейных лимфоузлов.
При выявлении аневризмы коронарных артерий достаточным считается наличие 3 из указанных диагностических признаков.
Лабораторная диагностика не дает специфических признаков болезни Кавасаки, однако совокупность выявленных изменений может стать дополнительным подтверждением правильности диагноза. В клиническом анализе крови определяется анемия, лейкоцитоз со сдвигом лейкоцитарной формулы влево, тромбоцитоз, значительное ускорение СОЭ. Биохимический анализ крови выявляет повышение иммуноглобулинов, серомукоида и трансаминаз, появление ЦИК. В анализе мочи может наблюдаться протеинурия и лейкоцитурия.
С целью диагностики сердечной патологии проводится ЭКГ, рентгенография органов грудной клетки, УЗИ сердца, ангиография коронарных артерий. По показаниям проводят люмбальную пункцию и исследование ликвора.
Лечение болезни Кавасаки
Ацетилсалициловая кислота. В современной медицине этот препарат назначается только при наличие строгих показаний. Однако в лечении болезни Кавасаки он входит в перечень необходимых медикаментов. Цель его применения — снижение риска образования тромбов и противовоспалительная терапия. После снижения температуры тела дозу ацетилсалициловой кислоты понижают до профилактической.
Антикоагулянты (варфарин, клопидогрел) назначаются для профилактики тромбообразования детям с диагностированными аневризмами сосудов. Кортикостероидная терапия при болезни Кавасаки не проводится, так как исследования показали, что она повышает риск коронарного тромбоза.
Прогноз болезни Кавасаки
Примерно в 20% случаев у перенесших болезнь Кавасаки детей сохраняются изменения стенок коронарных артерий, которые в отдаленном будущем могут привести к раннему появлению атеросклероза или кальциноза с последующей ишемией сердца, угрожающей развитием острого инфаркта миокарда. Факторами риска, ускоряющими развитие изменений со стороны коронарных артерий, являются артериальная гипертензия, гиперлипидемия, курение. В связи с этим пациенты с болезнью Кавасаки после выздоровления должны находиться под постоянным наблюдением кардиолога или ревматолога, раз в 3-5 лет проходить полное обследование сердца, включая ЭХО-ЭГ.
Геморрагический васкулит ( Аллергическая пурпура , Болезнь Шенлейн-Геноха , Капилляротоксикоз )
Геморрагический васкулит — системное асептическое воспаление сосудов микроциркуляторного русла с преимущественным поражением кожи, суставов, желудочно-кишечного тракта и почечных клубочков. Протекает с явлениями геморрагической или уртикарной сыпи, артралгиями, абдоминальным болевым синдромом, гематурией и почечной недостаточностью. Диагностика основана на клинических симптомах, лабораторных данных (анализ крови, мочи, коагулограмма), исследовании органов ЖКТ и почек. Основой лечения васкулита является терапия антикоагулянтами, ангиагрегантами. В тяжелых случаях применяется экстракорпоральная гемокоррекция, глюкокортикоидная терапия, противовоспалительное, цитостатическое лечение.
МКБ-10
Геморрагический васкулит (ГВ, болезнь Шенлейн-Геноха, аллергическая пурпура, капилляротоксикоз) относится к наиболее распространенным на сегодняшний день геморрагическим заболеваниям. По сути своей он является аллергическим васкулитом поверхностного характера с поражением мелких артериол, венул, а также капилляров. В Международной классификации болезней (МКБ) заболевание имеет название "аллергическая пурпура". Болезнь Шенлейн-Геноха встречается в основном в детском возрасте - от 5 до 14 лет. Средняя распространенность среди детей этого возраста составляет 23-25 случая на 10 тыс. Наиболее подвержены заболеванию лица в возрасте 7-12 лет. У детей до 3 лет известны лишь отдельные случаи возникновения пурпуры.
Причины
Этиологические аспекты изучены не до конца, известно лишь, что в большинстве случаев патология носит инфекционно-аллергическую природу. Существует сезонная зависимость ‒ наибольшая заболеваемость регистрируется в сырое и холодное время года. Многолетние наблюдения позволили выявить общие триггерные факторы, предшествующие развитию клинических проявлений. К их числу относят:
Во многих наблюдениях причинный фактор, вызвавший возникновение васкулита, установить не удается. Ряд авторов высказывает предположение, что воздействие провоцирующих факторов приводит к развитию геморрагического васкулита лишь в тех случаях, когда оно осуществляется на фоне генетической предрасположенности организма к гиперергическим иммунным реакциям.
Патогенез
В основе механизма развития геморрагического васкулита лежит образование иммунных комплексов и повышение активности белков системы комплемента. Циркулируя в крови, они откладываются на внутренней поверхности стенки мелких сосудов (венул, артериол, капилляров), вызывая ее повреждение с возникновением асептического воспалительного процесса. Воспаление сосудистой стенки в свою очередь приводит к повышению ее проницаемости, отложению в просвете сосуда фибрина и тромботических масс, что обуславливает основные клинические признаки заболевания — кожно-геморрагический синдром и микротромбирование сосудистого русла с поражением ЖКТ, почек, суставов.
В клиническом течении капилляротоксикоза различают острую фазу (начальный период или обострение) и фазу стихания (улучшение). По преобладающим симптомам заболевание классифицируют на следующие клинические формы: простую, ревматоидную (суставную), абдоминальную и молниеносную. В соответствии с характером течения различают острый (до 2-х мес.), затяжной (до полугода) и хронический ГВ. По тяжести клинических проявлений выделяют васкулит:
- Легкой степени. Отмечается удовлетворительное состояние пациентов и необильный характер сыпи, артралгии.
- Средней степени. Состояние больного средней тяжести, высыпания обильные, артралгии сопровождаются изменениями в суставах по типу артрита, отмечаются периодические боли в животе и микрогематурия.
- Тяжелой степени. Имеет место тяжелое состояние больного, сливные обильные высыпания с некротическими участками, ангионевротические отеки, нефротический синдром, наблюдается макрогематурия и желудочно-кишечные кровотечения, возможно развитие острой почечной недостаточности.
Симптомы
Для клиники аллергической пурпуры типично острое начало с повышением температуры до субфебрильных или фебрильных цифр. Однако возможно отсутствие подъема температуры. Кожный синдром отмечается в самом дебюте заболевания и наблюдается у всех больных. Он характеризуются диффузными пятнисто-папулезными геморрагическими элементами различной величины (чаще мелкими), не исчезающими при надавливании. В некоторых случаях наблюдается уртикарная сыпь. Высыпания обычно располагаются симметрично на коже голеней, бедер и ягодиц, в области крупных суставов, реже — на коже рук и туловища. Обильность высыпаний часто коррелирует с тяжестью васкулита. При наиболее тяжелом его течении в центре некоторых элементов сыпи развивается некроз и образуется язва. Разрешение сыпи заканчивается длительно сохраняющейся гиперпигментацией. При хроническом течении ГВ с частыми рецидивами на коже после исчезновения сыпи возникает шелушение.
Суставной синдром развивается у 70% пациентов. Поражения суставов могут носить кратковременный характер в виде легкой артралгии или сохраняться в течение нескольких дней с выраженным болевым синдромом, сопровождающимся другими симптомами артрита (покраснение, отечность) и приводящим к ограничению движений в суставе. Типичным является летучий характер поражения с вовлечением преимущественно крупных суставов, чаще коленных и голеностопных. Суставной синдром может появиться в начальном периоде васкулита или возникнуть позже. Зачастую он имеет преходящий характер и никогда не приводит к стойкой деформации суставов. Абдоминальный синдром может предшествовать кожно-суставным проявлениям или сопутствовать им. Он проявляется болями в животе различной интенсивности - от умеренных до приступообразных по типу кишечной колики. Пациенты часто не могут указать точную локализацию боли, жалуются на нарушения стула, тошноту и рвоту. Абдоминалгии могут появляться несколько раз в течение суток и проходят самопроизвольно или в первые несколько дней лечения.
Почечный синдром возникает у 25-30% пациентов и проявляется признаками хронического или острого гломерулонефрита с различной степенью гематурии. У ряда больных возникает нефротический симптомокомплекс. Поражение других органов при геморрагическом васкулите происходит довольно редко. Это может быть геморрагическая пневмония в виде кашля с прожилками крови в мокроте и одышки, кровоизлияния в эндокард, геморрагический перикардит, миокардит. Поражение сосудов головного мозга проявляется головокружением, раздражительностью, головной болью, эпиприступами и может вызвать развитие геморрагического менингита.
Осложнения
Поражение почек является самым стойким синдромом геморрагического васкулита, может осложняться злокачественным гломерулонефритом и хронической почечной недостаточностью. В тяжелых случаях аллергической пурпуры возникают желудочно-кишечные кровотечения, сопровождающиеся кровавой рвотой и присутствием крови в каловых массах, легочные кровотечения, кровоизлияния в вещество головного мозга (геморрагический инсульт). Массивные кровопотери могут привести к коллапсу и анемической коме. Осложнения абдоминального синдрома встречаются реже и представлены инвагинацией кишечника, перитонитом, тромбозом брыжеечных сосудов, некрозом части тонкого кишечника. Наибольшая частота летальных исходов регистрируется при молниеносной форме ГВ.
Проводя диагностику, ревматолог учитывает возраст пациента, изучает этиофакторы, сопоставляет клинические и лабораторные данные, исключает другие заболевания. При развитии почечного синдрома пациенту необходима консультация нефролога, при наличии абдоминальных болей - консультация гастроэнтеролога и хирурга. Диагностическая панель включает:
- Гематологические тесты. В общем анализе крови, как правило, отмечаются неспецифические признаки умеренного воспаления (лейкоцитоз и небольшое повышение СОЭ), увеличение количества тромбоцитов и эозинофилов. Биохимический анализ крови показывает увеличение иммуноглобулина А и СРБ. Большое диагностическое значение имеют результаты коагулограммы. Отсутствие в ней данных за нарушение свертывания при наличии клинических признаков геморрагического синдрома свидетельствует в пользу ГВ.
- Анализы мочи и кала. В анализе мочи выявляется гематурия, протеинурия, цилиндрурия. Пациентам с почечным синдромом показан мониторинг изменений в анализе мочи, проведение биохимии мочи, пробы Зимницкого, Нечипоренко. Для диагностики скрытого ЖКТ-кровотечения производят анализ кала на скрытую кровь.
- Инструментальную диагностику. С целью оценки состояния органов-мишеней выполняется УЗИ почек, УЗДГ почечных сосудов. Для исключения органических причин кровотечения из пищеварительного тракта и бронхов целесообразно проведение УЗИ брюшной полости, гастроскопии, бронхоскопии.
- Биопсию с гистологией. В тяжелых диагностических случаях показана биопсия кожи или почек. Гистологическое исследование биоптата выявляет характерные изменения: отложения иммуноглобулина А и ЦИК на эндотелии и в толще сосудистой стенки венул, артериол и капилляров; образование микротромбов; выход элементов крови за пределы сосуда.
Абдоминальную форму геморрагического васкулита следует дифференцировать от других причин, обуславливающих появление симптомов «острого живота»: аппендицита, пенетрации язвы желудка, острого холецистита, панкреатита, перфорации кишечника при язвенном колите др. Также необходимо исключить тромбоцитопеническую пурпуру, геморрагический синдром при инфекционных заболеваниях (геморрагических лихорадках, гриппе), лейкоз, ревматоидный артрит, болезнь Стилла, острый гломерулонефрит, системные васкулиты.
Лечение
В острой фазе геморрагического васкулита пациентам необходимо соблюдать постельный режим и гипоаллергенную диету, ограничить употребление жидкости и соли, исключить прием антибиотиков и других медикаментов, которые могут усиливать сенсибилизацию организма. Основные направления терапии зависят от клинических проявлений, поэтому их целесообразно рассматривать посиндромно:
- При любых синдромах. Основу базисной терапии при всех формах ГВ составляет назначение дезагрегантов (дипиридамола, пентоксифиллина) и активаторов фибринолиза (никотиновой кислоты). Препараты этих групп препятствуют агрегации тромбоцитов, улучшают микроциркуляцию и внутритканевую перфузию. Часто в базисную схему включают гепарин и другие антикоагулянты.
- При кожном синдроме. Терапия предполагает применение сульфасалазина, колхицина. Использование преднизолона до сих пор является спорным вопросом среди врачей. Возможно его назначение в тяжелых случаях ГВ. При отсутствии эффекта от терапии кортикостероидами препаратами запаса являются цитостатики.
- При суставном синдроме. Выраженные артралгии купируются проведением противовоспалительной терапии (индометацин, ибупрофен). Дополнительно могут назначаться производные аминохинолина (хлорохин).
- При почечном синдроме. Назначаются высокие дозы глюкокортикоидов, цитостатиков. Возможно использование иАПФ, антагонистов рецепторов ангиотензина II, введение нормального человеческого иммуноглобулина, проведение электрофореза с никотиновой кислотой и гепарином на область почек. В терминальной стадии ХПН требуется гемодиализ или трансплантация почки.
- При абдоминальном синдроме. Интенсивный болевой синдром служит показанием к внутривенному введению преднизолона, реополиглюкина, кристаллоидов. При развитии хирургических осложнений (перфорация, инвагинация кишки) применяется хирургическая тактика.
Тяжелое течение заболевания является показанием для проведения экстракорпоральной гемокоррекции (гемосорбция, иммуносорбция, плазмаферез). Многие авторы отмечают неэффективность антигистаминных препаратов в лечении ГВ. Однако их применение может быть оправдано у пациентов с аллергическим анамнезом. При связи заболевания с пищевой аллергией и наличием абдоминального синдрома дополнительно назначаются энтеросорбенты.
Прогноз и профилктика
Легкие формы геморрагического васкулита склонны к самопроизвольному излечению после первой же атаки заболевания - их прогноз благоприятен. При молниеносной форме смерть пациентов может произойти в первые несколько суток от начала заболевания. Чаще всего это связано с поражением сосудов ЦНС и возникновением внутримозгового кровоизлияния. Другой причиной летального исхода может стать тяжелый почечный синдром, приводящий к развитию уремии. В целях профилактики аллергического васкулита рекомендуется санация хронических инфекционных очагов ЛОР органов, дегельминтизация при глистных инвазиях, исключение контакта с известными аллергенами и бесконтрольного приема медикаментов.
Синдром (болезнь) Рейтера
Синдром (болезнь) Рейтера - ревматическое заболевание, характеризующееся сочетанным поражением урогенитального тракта (уретритом и простатитом), суставов (моно- или полиартритом) и слизистой глаз (конъюнктивитом), развивающимися последовательно или одновременно. В основе синдрома Рейтера лежит аутоиммунный процесс, вызванный кишечной или мочеполовой инфекцией. Диагностическими критериями являются связь с перенесенной инфекцией, лабораторное выявление возбудителя и характерных изменений крови, клинический симптомокомплекс. Лечение включает антибиотикотерапию инфекции и противовоспалительную терапию артрита. Синдром Рейтера имеет тенденцию к рецидивам и хронизации процесса.
В 80% случаев болезнь Рейтера атакует молодых мужчин от 20 до 40 лет, реже - женщин и исключительно редко - детей. Ведущим этиологическим агентом синдрома Рейтера служит хламидия - микроорганизм, способный к длительному паразитированию в клетках хозяина в виде цитоплазматических включений. Кроме того, синдром Рейтера может развиваться после перенесенного колита, вызванного шигеллой, иерсинией, сальмонеллой, а также провоцироваться уреаплазменной инфекцией. Предполагается, что перечисленные возбудители благодаря своей антигенной структуре вызывают определенные иммунологические реакции у генетически склонных лиц.
В течении синдрома Рейтера выделяют две стадии: инфекционную, характеризующуюся нахождением возбудителя в мочеполовом или кишечном тракте, и иммунопатологическую, сопровождающуюся иммунокомплексной реакцией с поражением конъюнктивы и синовиальной мембраны суставов.
Классификация синдрома (болезни) Рейтера
С учетом этиофактора различаются спорадическая и эпидемическая (постэнтероколитическая) формы заболевания. Спорадическая форма, или болезнь Рейтера, развивается после перенесенной мочеполовой инфекции; эпидемическая - синдром Рейтера - после энтероколитов различной этиологической природы (дизентерийных, иерсиниозных, сальмонеллезных, недифференцированных).
Течение болезни или синдрома Рейтера может быть острым (до 6 месяцев), затяжным (до года) или хроническим (длительнее 1 года).
Клиника синдрома (болезни) Рейтера
Для болезни (синдрома) Рейтера специфическими являются поражение урогенитального тракта, глаз, суставных тканей, слизистых и кожи. При болезни Рейтера первым манифестирует уретрит, сопровождающийся дизурическими расстройствами, скудным слизистым отделяемым, ощущениями дискомфорта и гиперемией в области наружной уретры. При бессимптомной клинике наличие воспаления определяется на основании увеличения числа лейкоцитов в мазке. Вслед за уретритом при синдроме Рейтера развивается глазная симптоматика, чаще имеющая форму конъюнктивита, реже - ирита, увеита, иридоциклита, ретинита, кератита, ретробульбарного неврита. Явления конъюнктивита могут быть мало продолжительными и слабо выраженными, незаметными для пациента.
Определяющим признаком синдрома Рейтера является реактивный артрит, который дебютирует спустя 1-1,5 месяца после урогенитальной инфекции. Для синдрома Рейтера типично асимметричное вовлечение суставов ног - межфаланговых, плюснефаланговых, голеностопных, коленных. Артралгии более выражены утром и по ночам, кожа в области суставов гиперемирована, в полости суставов образуется выпот.
Синдром Рейтера отличается последовательным лестничным (от проксимальных к дистальным) вовлечением суставов в течение нескольких дней. При урогенном артрите развиваются отеки, сосискообразные дефигурации пальцев; кожа над ними приобретает окраску синюшно-багрового цвета. При болезни Рейтера может развиваться тендинит, пяточный бурсит, пяточные шпоры, поражение крестцово-подвздошных суставов - сакроилеит.
Слизистые оболочки и кожные покровы при синдроме Рейтера поражаются у 30-50% пациентов. Характерны язвенные изменения слизистой рта (глоссит, стоматит) и полового члена (баланит, баланопостит). На коже появляются красные папулы, эритематозные пятна, очаги кератодермии - участки гиперемии кожи с гиперкератозом, шелушением и трещинами преимущественно на ладонях и стопах. При синдроме Рейтера возможно развитие лимфаденопатии, миокардита, миокардиодистрофии, очаговой пневмонии, плеврита, полиневритов, нефрита и амилоидоза почек.
При осложненной форме синдрома Рейтера развиваются дисфункции суставов, расстройства зрения, эректильные нарушения, бесплодие. В поздней фазе болезни Рейтера могут поражаться почки, аорта, сердце.
Диагностика синдрома (болезни) Рейтера
В ходе диагностики пациент с подозрением на синдром Рейтера может быть направлен на консультацию ревматолога, венеролога, уролога, офтальмолога, гинеколога. Общеклинические анализы при синдроме Рейтера выявляют гипохромную анемию, рост СОЭ и лейкоцитоз крови. В пробах мочи (трехстаканной, по Аддису-Каковскому и Нечипоренко) определяется лейкоцитурия. Микроскопия простатического секрета показывает увеличение лейкоцитов (>10) в поле зрения и снижение числа лецитиновых телец. Изменения биохимии крови при синдроме Рейтера характеризуются повышением α2- и β-глобулинов, фибрина, сиаловых кислот, серомукоида; наличием С-реактивного протеина, отрицательной пробой на РФ.
Цитологические исследования соскобов уретры, шейки матки, конъюнктивы, синовиального экссудата, спермы, секрета простаты с окрашиванием по Романовскому-Гимзе обнаруживает хламидии в виде внутриклеточных цитоплазматических включений. В диагностике синдрома Рейтера широко используется метод обнаружения ДНК возбудителя в биоматериале (ПЦР). В крови хламидийные и др. антитела выявляются с помощью серологических реакций - ИФА, РСК, РНГА. Специфическим признаком синдрома Рейтера является носительство антигена HLA 27.
В анализе синовиальной жидкости, взятой путем пункции сустава, определяются воспалительные изменения - рыхлость муцинового сгустка, лейкоцитоз (10-50×109/л), нейтрофилез свыше 70%, наличие цитофагоцитирующих макрофагов, хламидийных антител и антигенов, повышенная активность комплемента, РФ не выявляется. При рентгенографическом исследовании суставов выявляются признаки несимметричного параартикулярного остеопороза, уменьшения размеров суставных щелей, эрозивной деструкции костей стоп, наличия пяточных шпор и шпор пястных костей, тел позвонков, у трети пациентов - односторонний сакроилеит.
При диагностике синдрома Рейтера принимаются во внимание анамнестические сведения (связь заболевания с урогенитальной или кишечной инфекцией); наличие симптомов конъюнктивита, реактивного артрита, кожных проявлений; лабораторное подтверждение возбудителя в эпителиальных соскобах.
Лечение синдрома (болезни) Рейтера
Тактика лечения синдрома Рейтера предусматривает проведение антибиотикотерапии (для обоих половых партнеров), иммунокоррекции, противовоспалительного курса и симптоматической терапии. Антибиотикотерапия включает 2-3 последовательных курса (по 2-3 недели) препаратами из различных фармакологических групп: тетрациклинами (доксициклин), фторхинолонами (ломефлоксацин, офлоксацин, ципрофлоксацин) и макролидами (кларитромицин, азитромицин, эритромицин и др.). При хламидийной инфекции предпочтение отдается доксициклину. Одновременно с антибиотикотерапией назначается противогрибковые препараты, поливитамины, гепатопротекторы, протеолитические ферменты (панкреатин, трипсин, химотрипсин).
Иммунокоррегирующая терапия при синдроме Рейтера включает использование иммуномодуляторов (препаратов тимуса), адаптогенов, индукторов интерферона ( оксодигидроакридинилацетата натрия, акридонуксусная кислота в комбинации с N-метилглюкамином), а также УФОК, надвенной и внутривенной квантовой терапии. При тяжелых артралгических атаках и высокой активности воспаления проводится дезинтоксикационная и антигистаминная терапия. В целях дезинтоксикации при болезни Рейтера показана экстракорпоральная гемокоррекция - проведение плазмафереза, каскадной фильтрации плазмы и криоафереза.
Для подавления внутрисуставного воспаления при синдроме Рейтера используются НПВС (рофекоксиб, целекоксиб, нимесулид, мелоксикам), глюкокортикостероиды (бетаметазон, преднизолон), базисные препараты (сульфасалазин, метотрексат). При наличии внутрисуставного экссудата проводится лечебная пункция сустава с введением пролонгированных глюкокортикоидов (бетаметазона, метилпреднизолона). Местно накладываются компрессы с раствором диметилсульфоксида, обезболивающими и противовоспалительными мазями.
Стихание явлений острого артрита при синдроме Рейтера позволяет подключить физиотерапевтические сеансы фонофореза с протеолитическими ферментами, глюкокортикоидами, хондропротекторами; УВЧ, диатермию, магнитотерапию, лазеротерапию, массаж, грязелечение, сероводородные и радоновые ванны. В комплексе с терапией собственно синдрома Рейтера проводится лечение других экстрагенитальных очагов воспаления.
Прогноз и профилактика синдрома (болезни) Рейтера
Динамика течения синдрома Рейтера преимущественно благоприятная. У большей части пациентов через полгода заболевание переходит в стойкую ремиссию, что, однако, не исключает обострения болезни Рейтера много лет спустя. У четверти пациентов артрит переходит в хроническую фазу, приводя к дисфункции суставов, атрофии мышц, развитию плоскостопия. Исходом синдрома Рейтера может служить амилоидоз и другие висцеропатии.
Профилактика синдрома (болезни) Рейтера включает предупреждение кишечных и урогенитальных инфекций, проведение своевременной этиотропной терапии уретритов и энтероколитов.
Синдром Кавасаки - симптомы и лечение
Что такое синдром Кавасаки? Причины возникновения, диагностику и методы лечения разберем в статье доктора Похлебкиной Алевтины Алексеевны, педиатра со стажем в 6 лет.
Над статьей доктора Похлебкиной Алевтины Алексеевны работали литературный редактор Вера Васина , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Болезнь или синдром Кавасаки — это острое воспаление сосудов, которое встречается в основном у младенцев и детей в возрасте до пяти лет. Сопровождается лихорадкой, шелушением кожи и симптомами острого воспаления: гиперемией слизистой оболочки глазного яблока, покраснением слизистой оболочки полости рта, сыпью, увеличением шейных лимфатических узлов, покраснением и отёками кистей и стоп.
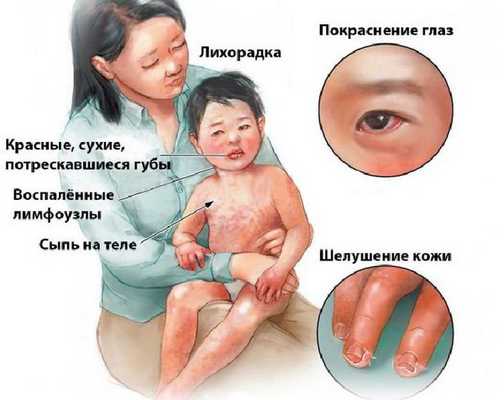
Впервые болезнь описана в Японии в 1967 году доктором Томисаку Кавасаки и впоследствии была признана во всём мире как важнейшее заболевание детского возраста [1] [2] . Синдром Кавасаки является наиболее распространённой причиной болезней сердца у детей в развитых странах.
Причины возникновения болезни Кавасаки до конца не изучены, долгое время к ним относили инфекции. На это указывало увеличение числа заболевших в конце зимы и весны и волнообразное географическое распространение эпидемий. Болезнь Кавасаки редко встречается у детей младше четырёх месяцев. Это позволяет предположить, что материнские антитела могут обеспечивать пассивный иммунитет [3] . Подозреваемыми инфекционными агентами были стафилококки, стрептококки, микоплазмы или хламидии, вирусы, такие как аденовирус, парвовирус или вирус Эпштейна ― Барра. Однако в носоглотке, ротоглотке, на коже или в кале больных возбудители выявлены не были [4] [5] [6] . Также было выдвинуто предположение об аутовоспалительном происхождении болезни Кавасаки [7] .
От болезни Кавасаки страдают все этнические группы, но особенно высок уровень заболеваемости в азиатских странах, среди японцев и корейцев, а также при их миграции в другие страны. Частота встречаемости болезни выше среди братьев, сестёр и близнецов, что предполагает генетический вклад в развитие синдрома [8] [9] . По данным на 2008 год, заболеваемость болезнью Кавасаки в Японии составляет 219 случаев на 100 000 детей, на Тайване — 69, в США — 9-19, в Великобритании — 8 [15] . Исследования выявили несколько генов, повышающих восприимчивость к заболеванию и её последствиям в различных этнических популяциях. К ним относятся FCGR2A, CD40, ITPKC, FAM167A-BLK и CASP3, а также гены, влияющие на ответ после введения внутривенного иммуноглобулина и способствующие образованию аневризмы (выпячиванию стенки артерии). На сегодняшний день вопрос генетического вклада в болезнь Кавасаки интенсивно изучается во всём мире.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Кавасаки
Большинство детей с болезнью Кавасаки нуждаются в медицинской помощи из-за продолжительной лихорадки. Основные симптомы заболевания [3] :
- Изменения со стороны кожи конечностей:
- эритема (покраснение) ладоней и подошв, иногда с болезненными отёками рук или ног, что может препятствовать движениям конечностей;
- чешуйчатое шелушение (отслаивание) эпителия пальцев рук и ног возникает в течение 2-3 недель после начала лихорадки;
- линии Бо (глубокие поперечные "канавки" на ногтях) могут появиться через 1-2 месяца после начала лихорадки.
- Полиморфная сыпь ― на покрасневшей коже появляются красные или фиолетовые бугорки, которые могут принимать различные формы.
- Изменения в ротоглотке:
- эритема, трещины, кровотечение и/или образование корок на губах;
- "клубничный язык" с выпуклыми грибовидными сосочками;
- рассеянное покраснение слизистой оболочки ротоглотки.
- Двусторонний, неэкссудативный (без появления эксудата — отделяемого), безболезненныйконъюнктивит наблюдается более чем у 90 % пациентов.
- Острая односторонняя негнойная шейная лимфаденопатия (увеличение лимфоузлов) с диаметром лимфатического узла не менее 1,5 см.
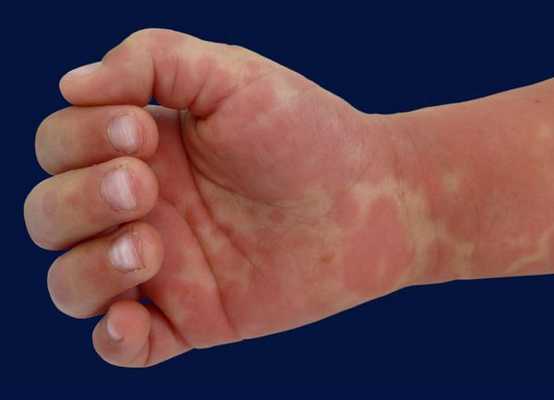
Немаловажный симптом ― высокая температура (часто 40 °C и выше) длительностью более пяти дней. Для постановки диагноза необходимы четыре диагностических критерия из пяти, указанных выше, плюс лихорадка. При аномалии коронарных артерий болезнь Кавасаки может быть диагностирована и при выявлении менее четырёх критериев [8] .
Раздражительность (беспокойство, плаксивость) является важным признаком, который почти всегда присутствует, хотя и не входит в диагностические критерии. Точный механизм раздражительности неясен, но это может быть связано с наличием неинфекционного менингита. Другие относительно распространённые состояния при болезни Кавасаки: артрит, пневмония, увеит, гастроэнтерит (заболевание желудочно-кишечного тракта), дизурия (расстройство мочеиспускания), отит (воспаление среднего уха).
Патогенез синдрома Кавасаки
Болезнь Кавасаки — это генерализованный системный васкулит, вовлекающий кровеносные сосуды по всему организму. Сосудистое воспаление наиболее выражено в коронарных артериях, но также может возникать в венах, капиллярах, мелких артериолах и крупных артериях. На ранних стадиях заболевания наблюдаются отёк эндотелия и субэндотелия сосуда. Отёки возникают из-за выраженной стимуляции цитокинового каскада и активацией эндотелиальных клеток, но внутренняя эластическая мембрана остается неповреждённой.
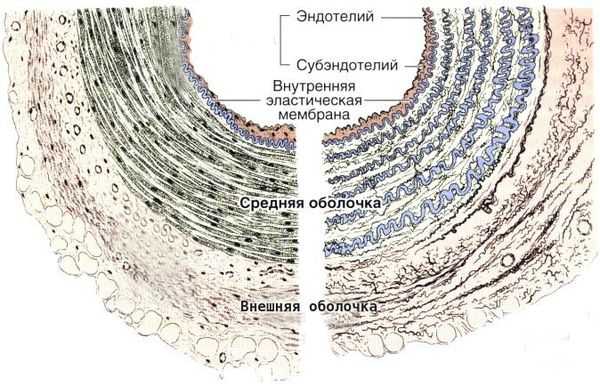
Цитокины — это белковые молекулы, вырабатываемые клетками для регуляции иммунного ответа. Они работают по принципу эстафеты: воздействие цитокина на клетку вызывает образование ею других цитокинов, этот процесс называется цитокиновым каскадом.
Воспалённые клетки вырабатывают различные цитокины и матриксные металлопротеиназы (ферменты, способные разрушать компоненты внеклеточного матрикса соединительных тканей), которые нацелены на эндотелиальные клетки. В результате происходит фрагментация внутренней эластической мембраны и повреждение сосудов.
Активное воспаление в течение нескольких недель или месяцев сменяется прогрессирующим фиброзом (разрастанием соединительной ткани) с образованием рубцов. В результате активного изменения сосудистой стенки и появления новых сосудов развивается стеноз (сужение просвета сосудов).
При повреждении мелких кровеносных сосудов к месту повреждения устремляются тромбоциты и образуют сгусток — тромб, закрывающий место дефекта сосуда.
Вследствие стеноза либо тромбоза просвет сосуда со временем сужается или закупоривается, что создаёт риск смерти от сердечно-сосудистых заболеваний, например от инфаркта миокарда.
Классификация и стадии развития синдрома Кавасаки
Существует две формы болезни Кавасаки:
- полная — наличие лихорадки не менее пяти дней и 4-5 клинических признаков, описанных выше;
- неполная ( "атипичная") — типичные клинические признаки заболевания отсутствуют; может наблюдаться, например, почечная недостаточность, которая не характерна для полной формы [3][10] .
Клинические проявления болезни Кавасаки меняются с течением времени. Условно выделяют три стадии заболевания: острую, подострую и выздоровление [3] [7] [10] . Некоторые авторы добавляют четвёртую хроническую стадию.
Острая стадия начинается с внезапного повышения температуры и длится примерно 7-14 дней. Лихорадка обычно сопровождается сильным всплеском и периодическими пиковыми температурами 39-40 °С и выше. Если лихорадка сохраняется, это может быть признаком рецидивирующей болезни Кавасаки. При таком течении заболевания высокая температура не снижается от приёма жаропонижающих препаратов и может сохраняться до 3-4 недель. После введения ВВИГ (внутривенного иммуноглобулина) лихорадка обычно проходит в течение 36 часов.
Подострая стадия начинается, когда лихорадка утихла, и продолжается до 4-6 недель. Отличительные признаки этой стадии: шелушение кожи пальцев, тромбоцитоз (количество тромбоцитов может превышать 1 млн/мкл) и развитие аневризмы. Если лихорадка держится более 2-3 недель возрастает риск сердечных осложнений. На этой стадии наиболее высок риск внезапной смерти.
Фаза выздоровления характеризуется полным исчезновением клинических признаков болезни, как правило, в течение трёх месяцев после начала заболевания. Эта стадия начинается с возврата к исходному уровню показателей: снижению количества тромбоцитов, лейкоцитов, СОЭ. На стадии выздоровления сердечные аномалии всё ещё могут быть выражены. Небольшие аневризмы в 60 % случаев разрешаются самостоятельно, но крупные могут расширяться, что создаёт риск инфаркта миокарда.
Хроническая стадия имеет клиническое значение только у тех пациентов, у которых развились сердечные осложнения. Она продолжается всю жизнь. В некоторых случаях недиагностированные разрывы аневризм во взрослом возрасте и эпизоды лихорадок неясного происхождения в детстве могут быть нераспознанными случаями болезни Кавасаки.
Осложнения синдрома Кавасаки
За последние пятьдесят лет болезнь Кавасаки стала одним из самых распространённых приобретённых пороков сердца у детей в мире.
Осложнения заболевания [11] :
- устойчивые формы болезни Кавасаки — постоянная или рецидивирующая лихорадка, невосприимчивая к терапии;
- сердечно-сосудистые патологии — расширение коронарных артерий, аневризмы, инфаркт миокарда;
- рецидивирующие формы болезни Кавасаки;
- другие системные осложнения болезни Кавасаки;
В последнее время появление аневризм коронарных артерий при болезни Кавасаки снизилось, благодаря лечению высокими дозами внутривенных иммуноглобулинов. Тем не менее в настоящее время в Японии около 0,2-0,3 % пациентов с болезнью Кавасаки имеют гигантские аневризмы коронарной артерии [12] . У пациентов с аневризмами более 8 мм в диаметре в течение первого года после начала болезни Кавасаки часто развивается острый инфаркт миокарда, что приводит к дисфункции левого желудочка или внезапной смерти [12] .
После болезни Кавасаки из-за длительного стеноза коронарной артерии может развиться ишемическая болезнь сердца. Тяжёлый локализованный стеноз из-за утолщения коронарной стенки после болезни Кавасаки может вызвать ишемию миокарда. Возникновение гигантской аневризмы часто свидетельствует о многососудистом поражении. Инфаркт миокарда у пациентов с гигантскими двусторонними аневризмами сильно влияет на исходы выживания на ранних и поздних стадиях после начала болезни Кавасаки.
К другим системным осложнениям болезни Кавасаки можно отнести анемию, гипоальбуминемию (снижение албуминов в крови), электролитные нарушения (особенно гипонатриемию — снижение натрия в крови), паралитический илеус (кишечную непроходимость), дисфункцию печени, холецистит, судороги, диарею, рвоту, дегидратацию и сердечную недостаточность, а также ятрогению, вызванную введением внутривенных иммуноглобулинов [11] .
Диагностика синдрома Кавасаки
Болезнь Кавасаки определяют на основании диагностических критериев, поскольку однозначных клинических признаков и специфических тестов не существует. При подозрении на болезнь Кавасаки важно рекомендовать госпитализацию, чтобы провести тщательную оценку и подтвердить диагноз [13] .
Типичная первоначальная лабораторная оценка может включать:
- общий анализ крови (ОАК);
- анализ электролитов;
- тестирование почечной функции;
- анализ ферментов печени, альбумина;
- определение скорости оседания эритроцитов (СОЭ);
- анализ крови на C-реактивный белок (CРБ);
- общий анализ мочи (ОАМ) [3] .
При острой стадии заболевания на ОАК часто выявляется анемия лёгкой и средней степени тяжести.
Во время подострой стадии распространён тромбоцитоз — повышение уровня тромбоцитов. Количество тромбоцитов начинает расти на второй неделе от начала заболевания и продолжает увеличиваться на третьей неделе. Повышение уровня маркеров воспаления, таких как СОЭ и СРБ, ― частое явление, но иногда они лишь незначительно возрастают.
Повышенные или умеренно высокие уровни сывороточных трансаминаз или гамма-глутамилтранспептидаз встречаются у 40-60 % пациентов, а лёгкая гипербилирубинемия (увеличение количества билирубина в крови) ― у 10 %. Гипоальбуминемия связана с более тяжёлым и длительным острым заболеванием. Анализ мочи может показывать пиурию (выделение гноя с мочой) у 80 % детей.
Для острой фазы болезни Кавасаки характерно нарушение липидного обмена, которое в конечном итоге приводит к снижению общего холестерина в сыворотке, особенно ЛПВП (липопротеинов высокой плотности), и увеличению триглицеридов.
При подозрении на болезнь Кавасаки выполняют эхокардиографию (ЭхоКГ). В дальнейшем исследование повторяют через 1-2 недели и через 5-6 недель после начала заболевания.
На электрокардиографии (ЭКГ) может определяться тахикардия, удлинённый интервал PR, изменение волны ST-T и снижение напряжения R-волн, указывающие на миокардит. Изменения волн Q или ST-T могут указывать на инфаркт миокарда.
Отдельной группе пациентов может потребоваться катетеризация сердца и ангиография. Ангиография сосудов позволяет детально исследовать артерии, но это может быть связано с большим риском осложнения во время манипуляции, особенно при выполнении в острой фазе заболевания. Коронарная компьютерная томографическая ангиография и магнитно-резонансная ангиография также будут полезны при оценке состояния и наблюдении за коронарными артериями.
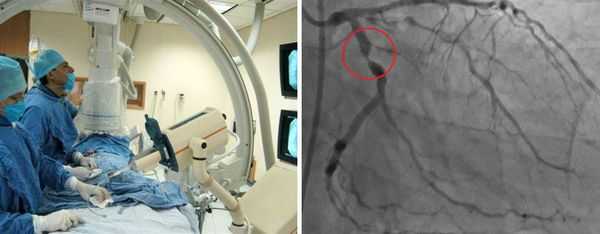
Ультразвуковое исследование показано при дисфункции того или иного органа. Пациентам с клиническими признаками менингита проводят люмбальную пункцию (взятие пробы спинномозговой жидкости).
Лечение синдрома Кавасаки
Основные цели лечения — подавить воспалительную реакцию и минимизировать риски развития аневризм коронарных артерий и других сердечных осложнений.
Все пациенты с болезнью Кавасаки должны быть госпитализированы для введения внутривенных иммуноглобулинов (ВВИГ) и аспирина, проведения эхокардиографии и наблюдения до устранения лихорадки [3] [13] .
В качестве "первой линии" лечения детей с болезнью Кавасаки применяют ВВИГ (внутривенные иммуноглобулины). Препараты наиболее эффективны, если назначены в течение первых 10 дней после начала лихорадки. В современной практике доза составляет 2 г/кг внутривенно в течение 10-12 часов.
Если после введения ВВИГ лихорадка сохраняется или возникает в течение 36 часов и позднее, то во многих из этих случаев рекомендуется повторное лечение ВВИГ в исходной дозе. Некоторые пациенты могут быть резистентными к действию ВВИГ, в таких случаях Американская кардиологическая ассоциация рекомендует пульс-терапию метилпреднизолоном, приём инфликсимаба, циклоспорина А, метотрексата и плазмаферез.
Большинство врачей используют аспирин в средних и высоких дозах в течение всего периода лихорадки, затем лекарство применяется в более низких дозах. Высокие дозировки требуются в острой фазе болезни для достижения противовоспалительного эффекта, в то время как более низкие дозировки препятствуют тромбообразованию в подостром периоде, когда существует риск развития аневризмы.
Прогноз. Профилактика
Без лечения смертность достигает 1 % и, как правило, случается в течение шести недель от начала заболевания. Длительная лихорадка увеличивает риск сердечно-сосудистых осложнений, в результате которых возможна внезапная смерть [14] .
При отсутствии ишемической болезни сердца прогноз для полного выздоровления хороший. Примерно две трети коронарных аневризм подвергаются регрессу в течение первого года. Гигантские аневризмы исчезают реже и требуют более интенсивного наблюдения и лечения.
Специфической профилактики не существует. Важными моментами при диспансеризации являются тромбопрофилактика, тщательное эхокардиографическое наблюдение за стенозами, закупорками коронарных артерий и ишемией миокарда. Каждый шесть месяцев необходимо проходить ЭхоКГ [15] .
Пациентам с тяжёлыми сердечными осложнениями может потребоваться катетеризация, шунтирование коронарной артерии или даже пересадка сердца. Успешное лечение требует эффективной совместной работы педиатров и кардиологов. Частота посещения доктора и приём лекарств зависят от тяжести заболевания. Поскольку дети, перенёсшие болезнь Кавасаки, имеют высокий риск развития осложнений со стороны сердечно-сосудистой системы, им может потребоваться наблюдение в течение жизни.
Читайте также: