Диагностика синдрома Айкарди на МРТ, КТ
Добавил пользователь Владимир З. Обновлено: 22.01.2026
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Ваша заявка принята!
Благодарим за обращение.
В ближайшее время с вами свяжется наш специалист.
Транзиторная ишемическая атака (ТИА): причины, симптомы, диагностика
Согласно эпидемиологическим данным примерно у 50-ти из 100 тысяч жителей европейских стран развивается транзиторная ишемическая атака (ТИА). Нозология относится к преходящим изменениям нарушения мозгового кровоснабжения, так как симптомы проходят, или значительно деградируют примерно через сутки после возникновения. Четко выявляется с помощью МРТ головного мозга и компьютерной томографии головы (эффективность ниже).
Статистика указывает на больший процент церебральной ишемии у женщин после 75 лет и мужчин после 65 лет. У молодых людей после 45 лет ТИА встречается редко.
Что такое транзиторная ишемическая церебральная атака
Длительность ишемических проявлений зависит от локализации патологии. в вертебробазилярной области (ВББ, шея и плечевое сплетение) продолжаются несколько часов. Эмболия, тромбоз передней и задней мозговой артерии обуславливает симптоматику до 24 часов.
Транзиторная церебральная ишемия - это состояние, которое некоторые врачи считают начальной стадии инсульта. Разница заключается лишь во временном промежутке сохранения клиники. Обе нозологические формы требуют тщательной диагностики состояния головного мозга в течение 60 минут после возникновения, так как промедление опасно для жизни пациента.
Преходящие приступы ишемии значительно повышают риск инсульта в течение 48 часов после первых проявлений.
Процент риска инсульта мозга после транзиторного церебрального приступа:
- На протяжении двух суток - 10%;
- Три месяца - 10%;
- Двенадцать месяцев - до 20%;
- Пятилетний срок - до 12%.
Учитывая статистику, важно понимать необходимость тщательной диагностики и правильного лечения ТИА на ранней стадии развития. Своевременное оказание помощи - это важный этап, но предотвратить инсульт после приступа удается только профилактическими процедурами.
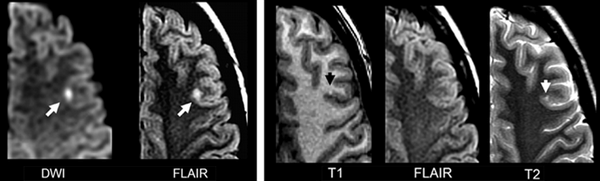
МРТ инсульта и транзиторной ишемической атаки
Классификация транзиторной церебральной ишемии по МКБ 10
Классификация ишемического приступа по международной классификации болезни десятого пересмотра сопровождается рядом нозологических форм:
- Транзиторная глобальная амнезия - «G45.4»;
- Двустороннее повреждение церебральных артерий - «G45.2»;
- Полушарный синдром сонной артерии - код «G45.1»;
- Преходящая слепота - «G45.3»;
- Вертебробазилярный синдром - «G45.0»;
- Мозговые атаки другие - «G45.8»;
- Ишемическая атака неуточненная - «G45.9».
Код МКБ 10 для транзиторных ишемических атак - «G45».
Определить нозологическую форму нарушения церебральной микроциркуляции следует сразу после поступления пациента, что позволит выбрать терапевтическую тактику, способы профилактики.
Симптом «преходящей слепоты» сопровождается появлением «заслонки» на одном глазу, которая возникает внезапно или формируется после раздражающего фактора - световая вспышка, пребывание под горячими лучами солнца. Сопровождать патологию могут мышечные судороги со стороны, противоположной очагу повреждения. Возможна потеря чувствительности кожных покровов.
Транзиторная амнезия характеризуется утратой памяти на кратковременные события. Во время приступа человек утрачивает ориентировку в окружающей обстановке, выполняет стереотипные действия.
Причины возникновения ишемической атаки
Распространенной причиной преходящей ишемии мозга являются микроэмболы, образующиеся из первичной атеросклеротической бляшки. Образования состоят из частиц холестерина, тромбоцитарных скоплений. После поступления внутрь мозговых артерий фрагменты блокируют кровообращение.
Другие причины транзиторного ишемического приступа:
- Васкулиты (ревматические, сифилитические, аутоиммунные);
- Гипертоническая болезнь (повышение артериального давления);
- Сахарный диабет;
- Заболевания свертывающей системы;
- Тромбангиит.
Неврогенное сужение сосудистого эндотелия возникает из-за раздражения стенки частицами атеросклеротической бляшки, кровяными сгустками. Патология кровоснабжения сопровождается отеком окружающих тканей, что повышает степень компрессии мозговой артерии.
Преходящую клинику формируют тромбоцитарные эмболы. Рыхлая структура образований способна распадаться.
Транзиторные приступы могут провоцироваться сосудистой недостаточностью, когда мозговые артерии обуславливают слабое поступление крови. Попадание внутрь артерии эмбола, тромба усиливает выраженность симптоматики. После устранение хронической недостаточности, разрушения тромбов внутримозговое кровообращение восстанавливается.
Симптомы церебрального ишемического приступа
Клиника ТИА зависит от локализации поврежденного церебрального сосуда.
Проявления обтурации вертебробазилярной артерии:
- Избыточное отделение пота;
- Внутриушной шум;
- Головокружение;
- Расстройства координации;
- Локальная амнезия (потеря памяти);
- Зрительные нарушения - двоение объектов, выпадение участков визуального образа, световые вспышки;
- Затылочные боли.
Распространенным проявлением вертебробазилярного синдрома является кратковременная потеря сознания, положительная проба Ромберга (невозможность дотрагивания пальцем до кончика носа).
Клиника полушарного синдрома сонной артерии
Специфичные признаки нозологии (код МКБ «G45.1»):
- Расстройства речи;
- Частичное или полное отсутствие зрения;
- Потеря тактильной чувствительности конечностей;
- Сниженный тонус мышц лица;
- Судорожные сокращения рук и ног.
Квалифицированный невролог по симптоматике сможет определить область повреждения мозга.
Чем проявляется тромбоз мозговых артерий
Симптоматика нарушения кровообращения по церебральным артериям:
- Нарушение двигательной активности конечностей с обеих сторон;
- Судорожные приступы;
- Двигательные и чувствительные расстройства с противоположной стороны;
- Выраженные нарушения речи.
Лучевая диагностика КТ и МРТ с контрастом верифицирует участки поврежденного кровотока шеи и головного мозга.
Чем отличается ТИА от инсульта
Стойкая окклюзия мозговых, позвоночных, сонных артерий обуславливает постоянный недостаток поступления кислорода к церебральной паренхиме. Последующие изменения окружающих тканей обуславливают гибель клеток. Некроз приводит к очаговым и общемозговым симптомам.
Вертебробазилярная недостаточность может провоцироваться дегенеративными изменениями шейного отдела позвоночника, при котором образуются задние костные разрастания области полулунных сочленений, приводящих к сужению позвоночной артерии.
Клиника недостаточности сонных артерий появляется до транзиторного приступа и характеризуется слабыми обморочными состояниями, которые нарастают по ходу прогрессирования остеохондроза, спондилеза, унковертебрального артроза.
По распространенности вертебробазилярная недостаточность встречается чаще эмболии каротидной артерии.
При ишемическом инсульте развиваются выраженные нарушения кровоснабжения мозга, сформированные внутренней окклюзией или внешней компрессией крупной артерии. Проявления сохраняются длительно и могут привести к летальному исходу.
Некоторые врачи называют преходящие церебральные ишемические приступы микроинсультом, так как примерно у половины части пациентов с нозологией развивается ишемический инсульт в течение года.
Ученые считаются ТИА предварительным компенсаторным механизмом перед последующими острыми ишемическими изменениями мозга. Появление преходящей атака способствует образованию коллатерального кровотока для предотвращения гипоксии.
Первые признаки транзиторной церебральной атаки
После возникновения первых проявлений требуется тщательная диагностика состояния человека. Возникновение любого из описанных принципов является показанием для обращения к врачу:
- Очаги дисциркуляторной энцефалопатии;
- Кратковременные нарушения сознания;
- Появление «пелены» перед глазами;
- Слепота на один глаз;
- Гемианестезия;
- Гемипарез;
- Расстройства чувствительности;
- Шум в ушах;
- Бледность лица;
- Синюшность кожных покровов;
- Затылочная боль;
- Вегето-сосудистые реакции;
- Динамическая атаксия;
- Нистагм.
Острое нарушение внутримозгового кровообращения (инсульт) можно предотвратить, если своевременно начать профилактику.

Восстановление после транзиторной атаки
У большинства людей после ТИА восстанавливаются практически все функции организма. Состояние обусловлено компенсацией недостатка микроциркуляции дополнительными системами организма:
- Увеличение частоты сердечных сокращений;
- Включение дополнительных шунтов за счет коллатеральных сосудов;
- Ускорение метаболических реакций.
Мнимое улучшение самочувствия является временным. Без профилактики и правильной реабилитации через некоторое время после ишемической транзиторной атаки появится инсульт.
Основная задача выяснить причины возникновения приступа с последующей коррекцией патологий:
- Антихолестериновая диета;
- Нормализации нарушений свертывающей системы;
- Коррекция обмена глюкозы;
- Симптоматическое лечение нарушений.
Клинические рекомендации реабилитационного периода включают здоровый образ жизни, отказ от вредных привычек, терапия вторичных заболеваний. Неврологи назначают препараты для нормализации мозгового кровоснабжения.
Синдром вертебробазилярной артериальной системы характеризуется кратковременными атаками, но восстановление после патологии невозможно. Большинство форм нарушения кровообращения ВББ прогрессирует медленно, так как обусловлены повреждением шейного отдела позвоночника.
Диагностика ТИА
После возникновения любого симптома человек должен госпитализироваться в неврологическое отделение. Специалисты европейских клиник способны в экстренном порядке сделать пациенту МРТ головы и КТ для отслеживания изменений головного мозга, диагностики ишемии или дифференцировки нозологических форм.
Визуализировать повреждение сосудов мозга позволяет МР или КТ-ангиография - процедуры контрастного обследования артерий после введения контраста внутрь вены. Одновременно с оценкой проходимости артериальной сети оценивают состояние сердечнососудистой системы:
- Холтеровское мониторирование;
- Эхокардиография;
- Электрокардиография.
Лабораторные способы диагностики:
- Определение антикардиолипиновых антител, волчаночного антикоагулянта, антитромбина III, протеина S и C, Д-димера, фактора Виллебранда;
- Изучение коагулограммы;
- Биохимический анализ.
После появления первых неврологических признаков церебрального ишемического приступа обязательно требуется консультация нескольких специалистов - окулиста, кардиолога, терапевта.
Диагностика синдрома Айкарди на МРТ, КТ
Российская медицинская академия последипломного образования;
Тушинская детская городская больница, Москва
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7): 95‑97
Милованова О.А. Синдром Айкарди. Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7):95‑97.
Milovanova OA. Aicardi syndrome. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2011;111(7):95‑97. (In Russ.).
Синдром Айкарди - врожденное мультисистемное заболевание неясной этиологии, характеризующееся тетрадой симптомов: эпилептическими приступами по типу инфантильных спазмов, частичным или полным отсутствием мозолистого тела, хориоретинальными лакунарными очагами, умственной отсталостью [1, 15, 21, 31, 32, 34]. Впервые заболевание было описано J. Aicardi в 1958 г. [4]. Всего в мире отечественными и зарубежными авторами описано 200 случаев синдрома Айкарди [1, 4, 11]. Считается, что синдром Айкарди - Х-сцепленное доминантное заболевание, приводящее к летальному исходу у лиц мужского пола [22, 26]. J. Holmes и соавт. [16] описали единственного мальчика с XXY-кариотипом. Некоторые исследователи предполагали, что все известные случаи заболевания являются результатами хромосомных мутаций, однако сведения о пораженных сибсах отсутствовали [11]. Не исключено, что эмбриональный мозаицизм при мутации генов может быть наследственно обусловленным [20]. H. Ropers и соавт. [26] выявили микроделецию в Хр22.3 у девочки с синдромом Айкарди. Предполагается, что патогенез данного заболевания связан с генетически детерминированными нарушениями на этапе нейрональной миграции. Морфологическое исследование головного мозга подтверждает множественные аномалии его развития, включающие полную или частичную агенезию мозолистого тела, гетеротопию коркового вещества мозга, аномалии строения извилин (чаще по типу микрогирии и гетеротопии), нарушения клеточной архитектоники.
Наиболее яркими клиническими проявлениями синдрома Айкарди являются неврологические расстройства. Эпилептические приступы в виде инфантильных спазмов доминируют у детей 1-го года жизни. По классификации О. Dulac и соавт. [12] различают флексорные, экстензорные, флексорно-экстензорные, тонические или миоклонические, симметричные или асимметричные, синхронные или асинхронные, с фокальным компонентом, серийные или единичные инфантильные спазмы. У детей с синдромом Айкарди чаще выявляются латерализованные флексорные инфантильные спазмы, сочетающиеся у части больных с другими видами эпилептических приступов (чаще фокальными). Установлено, что чем меньше возраст дебюта приступов при данном заболевании, тем тяжелее степень нарушения психомоторного развития [6, 8, 10].
У детей с синдромом Айкарди часто встречаются черепно-лицевые дисморфии (микро-, гидроцефалия, аномалии ушной раковины), реже - вертебральные и реберные мальформации.
Практически у всех детей с синдромом Айкарди выявляются нарушения психомоторного развития различной степени. В случаях с задержкой психомоторного развития, предшествующей дебюту эпилептических приступов, неврологический регресс в дальнейшем усугубляется [23]. Отсутствует интерес к окружающему миру, потерян контакт с ребенком, снижается дифференцировка эмоциональных реакций, практически прекращается предречевое развитие. Постепенно развивается двигательный дефект, в ряде случаев до степени тетрапареза центрального генеза с полным отсутствием развития двигательных навыков и установочных рефлексов. В 90% наблюдений нарушаются познавательные функции [25, 35].
В ряде случаев метод ультразвукового сканирования плода позволяет выявить наличие синдрома Айкарди на ранних стадиях гестационного развития.
При визуализационных методах - нейросонографии (НСГ), рентгеновской компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ) головного мозга - патогномоничным признаком синдрома Айкарди является агенезия мозолистого тела. Выделяют полную агенезию мозолистого тела, при которой отсутствуют все комиссуральные структуры вместе с фрагментами прозрачной перегородки, и частичную агенезию, которая в свою очередь подразделяется на агенезию ростральных и каудальных отделов мозолистого тела [29]. Чаще встречается агенезия каудальных отделов мозолистого тела, которая может сочетаться с изолированной дилатацией задних отделов боковых желудочков - кольпоцефалией. КТ-признаки агенезии мозолистого тела включают визуализацию межполушарной кисты, смещение вверх расширенного III желудочка и специфическое изменение формы тел боковых желудочков с увеличением расстояния между ними в виде симптома «ухвата» [7, 31]. Определяется расширение задних рогов боковых желудочков, своеобразный U-образный характер передних (фронтальных) рогов, удлиненная форма отверстия Монро. Кроме того, часто выявляются признаки атрофии вещества головного мозга в виде углубления и расширения конвекситальных борозд, расширения межполушарной и латеральной щелей, множественные пороки развития головного мозга.
S. Carney и соавт. [9] описали девочку с сочетанием признаков двух синдромов: Айкарди и де Морсье (септооптической дисплазии).
При записи паттерн-реверсивных зрительных вызванных потенциалов (ЗВП) у детей регистрируются ответы как на низко-, так и высокочастотные стимулы [1].
У детей раннего возраста с синдромом Айкарди, не имеющих нарушений со стороны оптических сред, макулы и диска зрительного нерва, острота зрения, определенная при помощи паттерн-реверсивных ЗВП или методом предпочтительного взора, может соответствовать возрастной норме [1, 19].
Синдром Айкарди необходимо дифференцировать от других мультисистемных заболеваний, характеризующихся церебральными аномалиями, изменениями диска зрительного нерва и сетчатки: туберозный склероз, септооптическая дисплазия, папилло-ренальный синдром, врожденные TORH-инфекции, перинатальные гипоксически-ишемические поражения, изолированная агенезия мозолистого тела, сочетанные и изолированные церебральные аномалии (полимикрогирия, гетеротопия, лиссэнцефалия) [1, 2, 9, 13].
В настоящее время лечение синдрома Айкарди носит исключительно паллиативный характер. Основная стратегия терапии - купирование эпилептических приступов у детей грудного возраста, т.е. инфантильных спазмов, которые зачастую резистентны к противосудорожной терапии. Применяют различные комбинации антиконвульсантов в максимально переносимых дозах. Для монотерапии применяют противоэпилептические препараты - вальпроат (конвулекс-капли) 30-100 мг/кг в сутки в 2-3 приема или вигабатрин (сабрил) 50-150 мг/кг в сутки (средняя суточная доза 100 мг/кг) в 2 приема. При неэффективности монотерапии переходят к политерапии: 1) вальпроат 30-80 мг/кг в сутки в 2-3 приема + вигабатрин (сабрил) 50-150 мг/кг в сутки в 2 приема; 2) вальпроат 30-80 мг/кг в сутки в 2-3 приема + этосуксимид 20-35 мг/кг в сутки в 2-3 приема; 3) вальпроат 30-80 мг/кг в сутки в 2-3 приема + фризиум 0,5-1,0 мг/кг в сутки (максимально 1 мг/кг в сутки) в 2 приема. Высокоэффективны кортикостероиды: синактен-депо, начиная с 0,1 мг внутримышечно 1 раз в сутки, с постепенным наращиванием по 0,1 мг 1 раз в 2-5 дней до дозы 1,0 мг в сутки. Продолжительность терапии синактеном-депо составляет 1-3 мес. Преднизолон (2-10 мг/кг в сутки), гидрокортизон (5-20 мг/кг в сутки) применяют перорально. Продолжительность лечения варьирует от 3 до 8 нед, возможна длительная терапия - 4-6 мес. Гормональную терапию целесообразно назначать в сочетании с базовыми антиконвульсантами [3].
Таким образом, практически у всех детей с синдромом Айкарди присутствует нарушение психомоторного развития различной степени. От возраста дебюта эпилептических приступов зависит степень тяжести когнитивного дефицита.
На ЭЭГ при инфантильных спазмах в ряде случаев регистрируются типичные изменения в виде гипсаритмии. Методы прижизненной нейровизуализации - НСГ, КТ, МРТ головного мозга - окончательно подтверждают агенезию мозолистого тела у детей с синдромом Айкарди. Хориоретинальные лакунарные очаги, увеличение экскавации диска зрительного нерва, сочетающиеся с поражениями постгеникулярных зрительных путей, в частности зрительной лучистости, определяемыми при радиологических исследованиях, являются офтальмоскопическими проявлениями данного заболевания.
Ранняя диагностика синдрома Айкарди, включающая невро-, радио- и офтальмологические исследования, позволяет достаточно полно представить диагностический профиль этого тяжелого и прогностически неблагоприятного заболевания.
Синдром Айкарди
Синдром Айкарди — это Х-сцепленное генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела с формированием хориоретинальных лакун и вариабельными эмбриональными аномалиями скелета. Типичной клинической картиной выступает триада признаков: инфантильные спазмы, задержка психического развития, нарушения зрения. Диагностика осуществляется при помощи электроэнцефалографии, церебральной МРТ, офтальмоскопии, изучения зрительных потенциалов, рентгенографии. Генетические методы диагностики пока не найдены. Лечение паллиативное с назначением комбинаций антиконвульсантов для постоянного приема, периодических курсов кортикостероидов.
МКБ-10



Общие сведения
Синдром Айкарди относится к редким расстройствам эмбрионального развития нервной системы. Впервые описан в 1965 году французским детским неврологом Жаном Айкарди как заболевание, характеризующееся следующей триадой признаков: агенезия мозолистого тела, офтальмологические расстройства, мышечные спазмы. Название синдрому в честь Ж. Айкарди было дано в 1972 г. Дальнейшее его изучение расширило список характерных клинических признаков, включив в них лицевой дисморфизм, умственную отсталость, аномалии позвоночного столба. Развитие методов нейровизуализации и генетических исследований позволило сделать диагностику более точной. По данным мировой статистики, всего насчитывается около 500 заболевших. Синдром связан с патологией Х-хромосомы, встречается исключительно у девочек. У плодов мужского пола возникают аномалии, не совместимые с жизнью.

Причины
Заболевание является генетической патологией, связанной с нарушением участка на Х-хромосоме. Считается, что оно имеет доминантное сцепленное с Х-хромосомой наследование. Теоретически вероятность рождения больной девочки от матери, страдающей синдромом, составляет 50%. Наличие патологической Х-хромосомы у мальчиков приводит к внутриутробной смерти.
На практике генетическая передача не зафиксирована, поскольку заболевшие девочки не могут иметь потомства в силу выраженных психомоторных расстройств. Все исследованные случаи синдрома Айкарди возникали de novo. Встречаются преимущественно спорадические варианты заболевания. Исключение составляет единственный известный случай у двух сестер. В связи с малым количеством заболевших, проведение достоверного исследования мутагенных триггеров затруднительно.
Патогенез
Патогенетические механизмы развития заболевания находятся в стадии изучения. Предполагается, что мутации генов обуславливают расстройство нейрональной миграции, происходящей в период между 12-й и 24-й неделями эмбрионального развития. В эмбриогенезе миграция неокортикальных нейронов на периферию приводит к образованию коры и подкорковых зон. Ее нарушения обуславливают дисгенезию мозолистого тела, аномальное формирование извилин и архитектоники мозга.
В норме в мозолистом теле проходят межполушарные проводящие пути, поэтому его дисэмбриогенез влечет нарушение связей между полушариями. Макроскопически в головном мозге выявляется микрогирия, субатрофия церебральной коры, перивентрикулярные кисты, гетеротопия, агенезия мозолистого тела. Микроскопически в сетчатке отмечается истончение слоев, уменьшение количества сосудов, гиперплазия пигментного эпителия.
Симптомы
Дети с синдромом Айкарди рождаются без видимых патологий, обычно в срок. В большинстве случаев беременность и роды протекают без осложнений. Заболевание манифестирует инфантильными спазмами в первые месяцы жизни. Спазмы представляют собой фокальные эпиприступы, при патологии Айкарди отличаются большой вариабельностью.
Возможны миоклонии, тонические судороги, ретропульсии, пропульсии. Наблюдаются симметричные и асимметричные, единичные и серийные судорожные приступы, типична тенденция к серийности. Флексорные спазмы отмечаются у 97% пациенток. В 42% случаях заболевания инфантильные спазмы сочетаются с другими видами эпиприступов, как фокальными, так и генерализованными. Фокальные припадки происходят с вовлечением преимущественно лицевой мускулатуры.
Задержка психомоторного развития характерна для всех больных синдромом. Отмечается обедненность эмоциональных реакций, утрачен контакт с окружающими, останавливается развитие навыков и предречевое развитие. Позже присоединяются моторные расстройства: геми- и тетрапарезы с повышением или снижением тонуса мышц, возможна спастичность. В отдельных случаях описана умеренная задержка развития, легкая степень олигофрении.
Офтальмологические аномалии вариабельны, включают: атрофию зрительного нерва, микроофтальмию, колобому. Возможна катаракта, пигментный ретинит. Клинически наблюдается значительное снижение зрительной функции. Со стороны желудочно-кишечного тракта характерна неустойчивость стула, проблемы с кормлением, гастроэзофагальный рефлюкс.
Среди челюстно-лицевых дисморфий описаны асимметрия лица, гипертелоризм, аномалии ушной раковины, выступающие резцы. У 33% пациенток отмечаются дисэмбриогенетические изменения позвоночника и ребер в виде полупозвонков, расщепления позвоночника, агенезии ребра, аномального реберно-позвоночного сочленения. В возрасте после 7 лет типично замедление роста, в пубертате — задержка полового развития.
Гемангиомы, невусы и другие дерматологические проявления описаны у 20% больных. В 7% случаев синдром Айкарди протекает с аномалиями конечностей: гипоплазией пальцев, синдактилией, каптодактилией.
Осложнения
Выраженная олигофрения, двигательные расстройства с первых месяцев жизни обуславливают глубокую инвалидность ребенка. Девочки нуждаются в постоянном уходе, половина из них неспособны к элементарному самообслуживанию. Нарушение мышечной иннервации при отсутствии постоянных реабилитационных мероприятий сопровождается гипотрофиями, в случаях спастичности — развитием контрактур.
Судорожный синдром с генерализованными припадками и кластерными приступами усугубляет неврологический дефицит, олигофрению. Позвоночные аномалии приводят к развитию выраженного сколиоза. Больные с синдромом Айкарди подвержены рецидивирующей пневмонии, имеют больший риск возникновения новообразований.
Диагностика
Внешний вид ребенка с лицевыми аномалиями, уменьшенным кончиком носа, латерально расположенными бровями позволяет врачу предположить генетическое заболевание. В неврологическом статусе показательны мышечная гипотония или односторонняя спастичность, снижение мышечной силы в конечностях, оживление рефлексов, снижение психического развития. Диагностика синдрома осуществляется совместно с генетиками, однако специфический генетический анализ еще не разработан. Назначаются следующие исследования:
- Электроэнцефалография. Наиболее типичным ЭЭГ-признаком синдрома Айкарди выступает гипсаритмия, появляющаяся после манифестиции инфантильных спазмов. Типично различие ЭЭГ-паттерна двух полушарий, феномен «расщепленного мозга». У ряда пациенток энцефалографическая картина имеет стертый характер. Наличие вариабельных судорожных припадков обуславливает полиморфизм изменений ЭЭГ.
- Офтальмоскопия. Выявляет наличие хориоретинальных лакун, визуализирующихся как белесоватые депегментированные участки сетчатки. Двусторонние лакуны расположены асимметрично, в 10-20% случаев очаги диагностируются только в одном глазу. При офтальмоскопии пациенток более старшего возраста диагностируется катаракта, колобома и/или атрофия диска зрительного нерва.
- Зрительные вызванные потенциалы (ЗВП). Исследуются для определения остроты зрения в младенческом возрасте и проведения дифдиагностики с другими офтальмологическими заболеваниями. У маленьких пациенток без изменений на глазном дне острота зрения может соответствовать возрастной норме.
- МРТ головного мозга. Визуализирует полное или частичное недоразвитие мозолистого тела, признаки корковой атрофии, пахигирию, расширение борозд. III желудочек расширен, вместе с боковыми желудочками смещен вверх, вокруг него выявляются церебральные кисты. Отмечается изменение формы латеральных желудочков, множественные области гетеротопии с отсутствием дифференцировки слоев коры, гидроцефалия.
- Рентгенография скелета. Необходима для диагностики костных аномалий, сопровождающих синдром Айкарди.
Перинатальная ДНК-диагностика не разработана. Выявление патологии возможно в ходе ультразвукового исследования беременной. По данным УЗИ 1 триместра возможна диагностика агенезии мозолистого тела. Позже выявляются микроцефалия, интракраниальные кисти, позвоночные аномалии, прочие пороки развития.
Дифференциальная диагностика
Необходимо дифференцировать патологию Айкарди от прочих ранних мультисистемных поражений, включающих сочетание дисгенеза церебральных структур и нарушения строения сетчатки. Первым симптомом септооптической дисплазии выступает горизонтальный нистагм, отсутствующий у пациентов с синдромом Айкарди. Отличительной чертой туберозного склероза служит наличие дерматологических симптомов: гиперпигментаций, ангиофибром, участков «шагреневой» кожи. Папилло-ренальный синдром протекает с поражением почек. Дифференциальная диагностика с врожденными TORH-инфекциями проводится на основании данных лабораторных обследований.
Следует отличать синдром Айкарди от заболевания Айкарди-Гутиера с аутосомно-рецессивным типом наследования. Последнее относится к прогрессирующим лейкодистрофиям детского возраста, характеризуется потерей приобретенных навыков, лимфоцитозом цереброспинальной жидкости, повышенным уровнем альфа-интерферона, кальцификатами в подкорковых структурах.
Лечение синдрома Айкарди
В основе терапии заболевания лежит паллиативная медикаментозная помощь и реабилитационные мероприятия. Базовой схемой является сочетание противосудорожных и гормональных фармпрепаратов, что позволяет добиться снижения частоты эпиприступов. Основными компонентами лечения являются:
- Антиконвульсанты. На первом этапе проводится монотерапия в максимально переносимой дозировке. Поскольку инфантильные спазмы при синдроме Айкарди отличаются стойкостью к противосудорожным препаратам, то приходится переходить к комбинированной схеме терапии, включающей 2-3 препарата.
- Глюкокортикоиды. Показали свою эффективность при внутримышечном введении в течение 1-3 месяцев. В ряде случаев по назначению врача возможна более длительное глюкокортикоидное лечение.
- Реабилитационная терапия. Направлена на поддержание двигательной активности, предотвращение мышечной атрофии, формирования контрактур при спастичности. Лечебные мероприятия включают массаж и специальную гимнастику, разработанные индивидуально с учетом имеющихся аномалий. Детям со средней степенью когнитивного дефицита показаны занятия с психологом, логопедом для максимально возможного развития навыков.
Прогноз и профилактика
Поскольку синдром Айкарди является генетическим заболеванием с неуточненными этиофакторами генных мутаций, то его специфическая профилактика затруднительна. Возможно проведение общих профилактических мероприятий, направленных на охрану женщины от вредоносных воздействий в период беременности.
1. Синдром Айкарди/ Милованова О.А.// Журнал неврологии и психиатрии им. С.С. Корсакова - 2011. - 111(7).
2. Синдром Айкарди/ Петухова Н.М., Павловец Л.П., Жакашева А.С.// Вестник Алматинского государственного института усовершенствования врачей. - 2010. - 1.
3. Aicardi syndrome: an epidemiologic and clinical study in Norway/ Lund, C., Bjørnvold, M., Tuft, M., Kostov, H., Rosby, O., Selmer, K. K.// Pediatric Neurology. - 2015. - 52(2).
4. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children/ Glasmacher, M. A., Sutton, V. R., Hopkins, B., Eble, T., Lewis, R. A., Park Parsons, D., & Van den Veyver, I. B. // Journal of child neurology. - 2007. - 22(2).
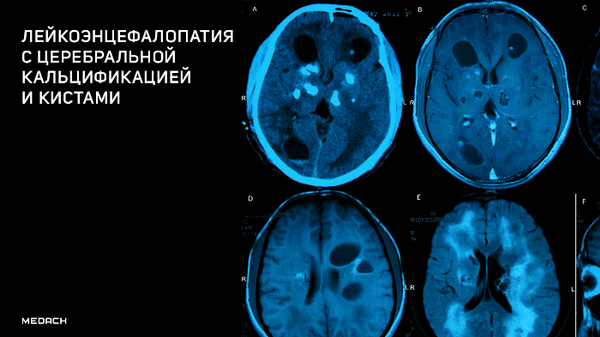
Лейкоэнцефалопатия c церебральной кальцификацией и кистами
Мальчик 10 лет, весом 23 кг, рожденный от неродственного союза (роды прошли без осложнений); жалуется на головные боли пульсирующего характера на протяжении 1 года, преимущественно в лобной и затылочной областях, почти непрерывные; боли усиливались при кашле и мешали пациенту спать и учиться. Мальчик был госпитализирован после обострения головной боли, что сопровождалось неоднократной рвотой и парциальными приступами в виде клонических подергиваний мышц левой половины лица. Объективное исследование не обнаружило отклонений от нормы.
Офтальмолог: не выявлено телеангиэктазий, экссудатов, ретинопатии, атрофии зрительного нерва или каких-либо других патологических признаков. Неврологическое исследование: ребенок в сознании, ориентирован, умеренно повышен мышечный тонус, оживленные глубокие сухожильные рефлексы, нерезко выраженный двусторонний положительный симптом Гордона, а также легкая ригидность мышц шеи и положительный симптом Кернига. Определяются мозжечковые симптомы: дисметрия (нарушение координации движений из-за утраты чувства расстояния, соразмерности и точности движений - прим.авт.), интенционный тремор, атактическая походка.
В анамнезе: туберкулезный лимфаденит в возрасте 2х лет, проводилось лечение противотуберкулезными препаратами.
Общий анализ крови, СОЭ, показатели функции почек и печени, уровни кальция, фосфата в плазме крови, щелочной фосфатазы, рентгенограмма грудной клетки и УЗИ органов брюшной полости были в пределах нормы.
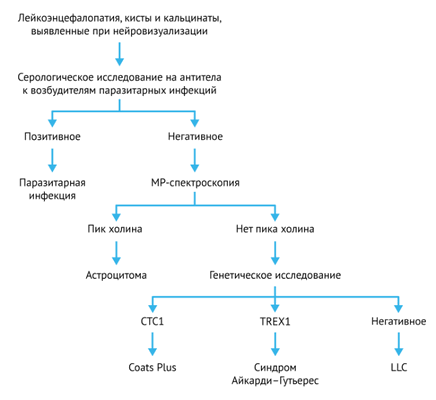
Рисунок 1 | Общий алгоритм дифференциальной диагностики
Серологические тесты не подтвердили эхинококкоз, токсоплазмоз, цистицеркоз, криптококкоз, цитомегаловирусную и ВИЧ-инфекцию.
Компьютерная томография показала признаки лакунарного черепа (определялись группы круглых, овальных или пальцевидных вдавлений на внутренней поверхности свода черепа, разделенные ребристыми выростами нормальной костной ткани в самых толстых частях лобной, теменной и верхней затылочной костей - характерный признак внутриутробного повышения внутричерепного давления или нарушения костеобразования - прим.пер.), а также обширные области интракраниальных кальцинатов, которые распространялись билатерально на глубокия* ядра мозжечка, границу белого и серого вещества, таламусы, область базальных ядер и капсулы.
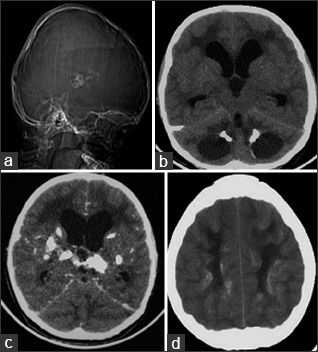
а: на топограмме, которая проводится для разметки сканирования, хорошо видны лакунарный череп и интракраниальные кальцинаты.
b: КТ без контрастного усиления показала двусторонние кальцинаты в зубчатом ядре мозжечка;
с: несимметричная двусторонняя обширная кальцификация в области базальных ядер и внутренней капсулы, таламусов и
d: границы серого и белого вещества
На МРТ головного мозга определялись обширные области гиперинтенсивного в Т2ВИ и FLAIR сигнала, которые затрагивали перивентрикулярные области белого вещества, оставляя интактными U-волокна и мозолистое тело с кистозными включениями и признаками обструктивной гидроцефалии. Обнаруженные ранее на КТ кальцинаты были гипоинтенсивны в Т2- и Т1ВИ и показывали «выпадение» МР-сигнала в GRE.
Клиническая и рентгенологическая картины были классическими для лейкоэнцефалопатии с кальцинатами и кистами (LCC, также называемая синдром Лабруна - прим.авт.), которая осложнилась развитием обструктивной гидроцефалии.
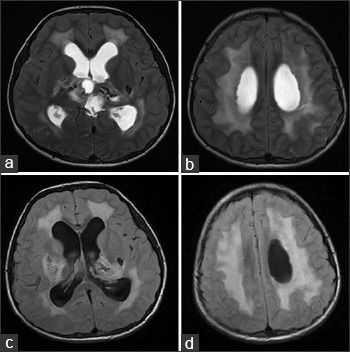
На аксиальных Т2ВИ (a, b) и FLAIR (с, d) томограммах можно увидеть обширные области гиперинтенсивности белого вещества мозга, что говорит о лейкоэнцефалопатии.
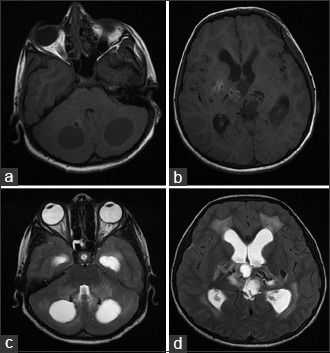
Аксиальные Т1 и Т2ВИ сканы демонстрируют кисты в обеих гемисферах мозжечка, расположенные над третьим желудочком в пинеальной области; обратите внимание на расширение боковых желудочков в результате обструктивной гидроцефалии.
Нейрохирургическая команда рекомендовала установку вентрикуло-перитонеального шунта, но родственники пациента отказались от инвазивных процедур. Пациенту было назначено лечение вальпроевой кислотой и ацетазоламидом, которые давали симптоматический эффект.
Области “выпадения” МР-сигнала на GRE в области базальных ядер, таламусов и ядер мозжечка соответствуют кальцинатам.
Сочетание обширных церебральных кальцинатов, изменений белого вещества мозга и кист были описаны Labrune с соавт. в 1996 году под названием LCC. Это крайне редкое состояние, всего в литературе было описано около 10 случаев; оно встречается у детей и взрослых; время появления симптоматики до 59 лет. Классическая для этой патологии клиника прогрессирующего неврологического дефицита у ребенка предполагает огромный список дифференциальных диагнозов, однако существуют характерные рентгенологические признаки, которые позволяют поставить диагноз почти безошибочно.
При дифференциальной диагностике стоит помнить о таких паразитарных инфекциях, как эхинококкоз, нейроцистицеркоз, криптококкоз.
При нейроцистицеркозе имеются множественные кистозные включения с вариабельной* точечной кальцинацией; желатинозные псевдокисты и паренхиматозные кальцинаты описаны у ВИЧ-инфицированных больных при криптококкозе, однако результаты серологических исследований не подтверждали этих нозологий, тем более, для них не характерны проявления лейкоэнцефалопатии (поражения белого вещества).
У нашего пациента наблюдалось повышенное внутричерепное давление и связанный с кистой масс-эффект, которые являются основными проявлениями LCC.
Данные нейровизуализации тесно связаны с патогенезом. Типично заболевание проявляется двусторонними асимметричными кальцификатами в подкорковых ядрах и ядрах мозжечка, диффузным поражением белого вещества, множественными кистами разного размера с признаками накопления контраста в их стенках, и, изредка, кровотечением в кисты или паренхиму мозга. МР-ангиография обычно без отклонений от нормы, перфузионное исследование определяет гиперперфузию в стенках кист, что вместе с возможными кровотечениями наталкивает на мысль о сосудистых нарушениях.
МР-спектроскопия, которая была проведена в некоторых случаях LCC, не выявила лактата и показала снижение пиков холина и NAA в областях пораженного белого вещества; эти изменения соответствуют содержанию воды в областях лейкоэнцефалопатии, в то время как от демиелинизирующего процесса ожидается высокий уровень холина. Возможно, отек белого вещества мозга связан с нарушениями ГЭБ. При проведении МР-спектроскопии содержимого кист типичных метаболитов для паренхимы мозга в них выявлено не было. При гистологическом исследовании в большинстве случаев обнаруживались волокна Розенталя (также встречающиеся при болезни Александера). Эти волокна, связанные с мутациями в гене, кодирующем глиальный фибриллярный кислый белок. (GFAP) представляют собой цитоплазматические включения в астроцитах, которые содержат белок промежуточной нити GFAP и небольшие белки теплового шока. Накопление волокон Розенталя может препятствовать нормальной функции астроцитов.
Помимо волокон Розенталя, наиболее частыми находками являются бледность миелина, ангиоматозные изменения сосудов мозга, микрокальцинаты и отложения гемосидерина.
Этиология заболевания в настоящее время не ясна. Кроме мутаций в гене GFAP, сообщалось также о связи мутаций в гене SNORD118 с данной нозологией.
Для окончательного подтверждения диагноза может проводиться биопсия, однако в большинстве случаев от этой инвазивной процедуры бывает больше вреда, чем пользы. Потому диагностика и дифференциальная диагностика в большей мере опираются на данные нейровизуализации, на основании данных которой можно сразу отсеять несколько схожих генетически обусловленных патологий.
Так, болезнь Фара, синдром Стерджа-Вебера и MELAS могут обусловливать интракраниальные кальцификаты, но без лейкоэнцефалопатии и кист. Синдром Кокейна представляет собой внутричерепные кальцификаты и диффузную лейкоэнцефалопатию, но без кист. Болезнь Гиппель-Линдау приводит к кистам мозжечка, но без кальцификатов и лейкоэнцефалопатии. Болезнь Александера представляет собой лейкоэнцефалопатию и кисты, но без кальцификации. Метахроматическая лейкодистрофия и адренолейкодистрофия приводят к диффузному поражению белого вещества мозга без кист и кальцификации.
Только LCC, Coats Plus синдром, синдром Айкарди-Гутьера, внутричерепная паразитарная инфекция и некоторые астроцитомы могут проявляться лейкоэнцефалопатией, кальцификатами и кистами. Coats Plus является системным заболеванием, связанным с мутацией гена CTC1. О внутричерепной паразитарной инфекции следует говорить после серологического подтверждения. Синдром Айкарди-Гутьера связан с мутацией TREX1. Астроцитомы предполагают повышение пика холина при МР-спектроскопии.

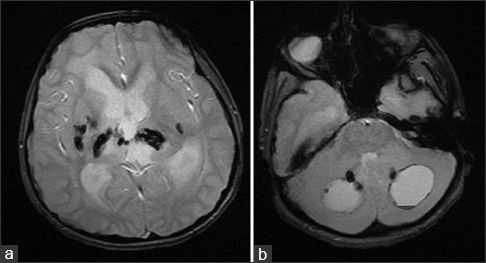
Аксиальные КТ-сканы (A, B) показывают двусторонние кальцинаты в области таламусов и хвостатых ядер, а также множественные кисты в обеих гемисферах мозга. Т2ВИ томограмма (С) показала диффузный патологический МР сигнал от белого вещества при сохранном сером веществе мозга. МРТ после введения контрастного препарата (D) показала, что киста в левой лобной доле демонстрирует неоднородное кольцевидное контрастное усиление.
МР-спектроскопия (E): с нижение пиков NAA и холина в областях измененного белого вещества. Перфузионная МРТ (F) показала гиперперфузию в стенке кисты. Т2ВИ через 5 месяцев после болезни (G): крупная киста левой лобной доли уменьшилась, а меньшая по размеру киста левой лобной доли, наоборот, увеличилась.
Miao Wang и соавт. предложили следующую схему диагностики для случаев сочетания поражения белого вещества с интрацеребральными кальцификатами и кистами.
КТ или МРТ брюшной полости
В нашей клинике вы можете пройти МРТ органов брюшной полости с выдачей медицинского заключения. По результатам МРТ исследования врач ставит точный диагноз и объективно оценивает состояние прошедшего обследование в клинике пациента.

Потребность в неинвазивных методах диагностики привела к развитию технологий, основанных на рентгеновских лучах и явлении ядерного резонанса. Ценную информацию о состоянии органов брюшной полости дает компьютерная томография и магнитно резонансная томография. Использование каждого из этих методов имеет свои показания и противопоказания.
Когда показана компьютерная томография
На рентгеновских томограммах, сделанных с помощью компьютера, хорошо визуализируются полые органы. В особенности, с применением контрастирующих веществ. Это отличная альтернатива эндоскопическому исследованию желудка, кишечника, и особенно желчного пузыря, недостижимого с помощью эндоскопа.
На КТ четко визуализируются кости, что предоставляет ориентиры хирургам, когда предстоит хирургическая операция. КТ дает возможность подготовиться с привязкой к индивидуальным особенностям организма, например, узнать, на каком уровне находится червеобразный отросток слепой кишки.

В каких случаях показана магнитно резонансная томография
В свою очередь, МРТ органов брюшной полости и забрюшинного пространства идеально подходит для визуализации плотных тканей, богатых водой. Водород, которого больше всего в составе воды, интенсивно откликается на поля, создаваемые аппаратом.
На экране появляются резкие контрасты между участками с разным содержанием воды. Видны границы печени, поджелудочной железы, почек, также небольших полостей внутри массивных структур. МРТ заменяет УЗИ, и технически проще осуществляется. Не нужно, чтобы специалист высокой квалификации долго исследовал пациента ультразвуком, выбирая наилучшее положение излучателя, и затем правильно интерпретировал результаты.
Противопоказания к рентгеновскому исследованию
- Необходимость делать повторные или регулярные снимки. Каждый снимок дает лучевую нагрузку в области живота, что совсем не полезно для здоровья.
- Беременность. Пока плод находится в организме матери, противопоказаны любые радиоактивные воздействия.
Противопоказания к магнитно резонансному исследованию
- Наличие металлических протезов в исследуемой части тела.
- Чрезмерное увеличение массы тела (выраженная тучность).
- Нервные и психические нарушения, в результате которых пациент не может оставаться неподвижным внутри аппарата.
Сравнение методик и выбор оптимальной для пациента
В большинстве случаев предпочтительно провести МРТ. Речь идет о мягких тканях, заполняющих брюшную полость, и очевидно, что в данной ситуации КТ менее информативна. Даже полые внутренние образования, как петли кишок и желчевыводящие пути, неплохо контрастируются на МРТ с использованием специальных контрастирующих веществ.
Достаточно взглянуть на снимки внутренних органов, выполненные разными методами, чтобы увидеть существенную разницу в уровне детализации. Некоторые структуры брюшного пространства одинаково хорошо видны при использовании соответствующего контраста, отсутствуют серьезные различия на КТ или МРТ. Но другие, в частности, расположенные за брюшиной, на КТ едва заметны.

Однако, КТ завершается намного быстрее. В срочных случаях достаточно нескольких минут, чтобы определить, к примеру, расположение инородного тела, проглоченного ребенком. Металл или пластик будет хорошо заметен на КТ даже без предварительного введения контраста.
Подготовка пациента
Когда проводится МРТ или КТ брюшной полости, необходима подготовка к обследованию. При контрастировании внутрь или в сосуды вводится контрастное вещество. Также следует подготовить пищеварительный тракт к исследованию брюшной полости, чтобы каловые массы и избыточные объемы газов не мешали диагностировать патологические изменения.
При обращении в медицинские центры пациентам подробно разъясняют, какой диеты придерживаться. Обычно за 2-3 дня следует уменьшить количество принимаемой пищи и употреблять продукты, не приводящие к метеоризму или запору. Подробный план подготовки к МРТ вам предоставит медицинский персонал, то же самое касается КТ.
Что диагностируют магнитным резонансом?
Поскольку брюшная полость обширная и состоит из нескольких отделов, на томографе выявляются заболевания многих систем: пищеварительной, мочеполовой, эндокринной. Также проведение МРТ бывает полезным при поражении крупных сосудов, хотя и КТ в данном случае информативна (с контрастированием).
Отдельную категорию исследований составляет использование магнитного поля для диагностирования опухолей. С помощью МРТ эффективно выявляют рак желудка, печени, почек, метастазы опухолей с первичным источником за пределами брюшной полости. Многие опухолевые процессы крайне сложно или практически невозможно выявить на КТ, поэтому очевидно, какой метод диагностики необходимо использовать.
Как проходит магнитно резонансное исследование?
Пациента помещают на выдвижной стол огромного устройства, внутренняя часть которого напоминает цистерну. Положение лежа на спине, чтобы брюшная стенка находилась сверху. Если лечащим врачом указана другая поза, обслуживающий персонал фиксирует пациента согласно назначению. Затем стол задвигается внутрь аппарата.
На стенке находится кнопка экстренной связи, нажимая которую, пациент может срочно вызвать помощь. Лежать следует спокойно, пока проходит МРТ, нельзя совершать какие-либо движения. Оператор передает по громкой связи указания. Иногда необходимо задержать дыхание, максимум на несколько секунд. Это позволяет получить четкие томограммы, не смазанные дыхательными движениями.
Когда завершается диагностическая процедура, стол снова выезжает, пациент встает самостоятельно или переносится на носилки (каталку). Все абсолютно безболезненно, отсутствуют какие-либо неприятные ощущения.
Что видно на томограмме?
Как и в случае КТ, производится сложная компьютерная обработка изображений, полученных на МРТ. В итоге получается очень четкая картина патологий, затронувших брюшную стенку, внутрибрюшинное пространство и структуры организма, находящиеся дальше.
Врач сразу видит, какие органы поражены, и может наглядно показать пациенту или родственникам. Изображение с МРТ гораздо понятнее неспециалистам, чем КТ, или, тем более, полученное при УЗИ.
Наглядность помогает лучше обосновать необходимость оперативного вмешательства, получить согласие на хирургическое лечение. Снимки с МРТ передаются пациенту в цифровом виде, скажем, как записанные на флешке файлы. Впоследствии могут быть распечатаны на домашнем принтере и просмотрены на экране компьютера.
В нашей клинике делают МРТ в строгом соответствии со стандартами, получая качественные результаты, которые легко интерпретировать любому специалисту. При наличии противопоказаний вы можете пройти КТ или УЗИ. Вас осмотрит опытный врач, выявит показания и направит на исследование, дающие лучшие результаты.
Читайте также: