Диагностика врожденной мышечной дистрофии на МРТ, КТ
Добавил пользователь Валентин П. Обновлено: 01.02.2026
Врожденные миопатии - группа заболеваний, проявляющихся с рождения или в раннем детском возрасте снижением силы мышц, их гипотонией, ослаблением или отсутствием сухожильных рефлексов. Отличаются от прогрессирующих мышечных дистрофий доброкачественным течением. В ряде случаев симптомы дефицита мышечной системы остаются стабильными на протяжении всей жизни, иногда незначительно прогрессируют, возможно, даже регрессивное течение.
По клинической картине распознать эти заболевания чрезвычайно трудно, почти у всех больных имеется мышечная слабость, которая может быть генерализованной или преобладать в проксимальных отделах конечностей и мышцах плечевого и тазового пояса, имеются диффузная мышечная гипотрофия и гипотония, сухожильные рефлексы резко снижены или отсутствуют, иногда развиваются контрактуры.
При выраженной степени расстройств обычно ставится диагноз «floppy baby» (вялый ребенок).
Врожденная мышечная дистрофия - мышечные заболевания, имеющиеся с рождения или возникающие вскоре после рождения. Диагностика этих заболеваний может быть основана на следующих критериях:
- гипотония, слабость или артрогрипоз представлены с рождения;
- мышечная биопсия обнаруживает признаки миопатии (изменение размеров волокон, дегенерация волокон среднего размера, замещение мышечных волокон жировой тканью и коллагеном) и исключает денервацию;
- другие формы миопатии со специфическими клиническими признаками у новорожденных исключены.
Протокол "Врожденная миопатия"
Код по МКБ-10: G71.2
Врожденная мышечная дистрофия:
- со специфическими морфологическими поражениями мышечного волокна.
Диспропорция типов волокон
- немалиновая (болезнь немалинового тела).

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
1. Врожденные миопатии.
2. Болезнь центрального стержня.
3. Врожденная миопатия с диспропорцией типов волокон.
4. Миотубулярная (центронуклеарная) миопатия.
5. Острая миотубулярная миопатия.
6. Хроническая миотубулярная миопатия.
7. Немалиновая миопатия.
Диагностика
Диагностические критерии
Жалобы и анамнез: заболевание проявляется с рождения или в раннем детском возрасте; задержка в моторном развитии, гипотрофия, мышечная слабость, снижение тонуса и мышечной силы, нарушение походки.
Физикальные обследования
Болезнь центрального стержня
Наследуется по аутосомно-доминантному типу, характеризуется специфическими морфологическими изменениями, при которых в мышечных волокнах в центральной части имеются участки, заполненные компактными миофибриллами - "стержни". Диаметр волокон сохраняется, иногда он даже несколько увеличен.
Клиническая картина характеризуется более или менее значительной мышечной слабостью, преобладающей в проксимальных отделах конечностей, особенно нижних, и диффузной мышечной гипотонией.
Типично отставание в двигательном развитии при нормальном интеллекте. Мышечные атрофии выражены умеренно, сухожильные рефлексы сохранены или нерезко снижены.
Заболевание, как правило, не прогрессирует, к пубертатному периоду возможен регресс симптомов. Продолжительность жизни не ограничивается. Существуют субклинические формы.
Болезнь центрального ядра (миотубулярная миопатия)
Типы наследования: аутосомно-доминантный, аутосомно-рециссивный, рециссивный, сцепленный с Х-хромосомой. Эти обстоятельства говорят о генетической гетерогенности миотубулярной миопатии. Морфологические изменения при миотубулярной миопатии характеризуются уменьшением диаметра мышечных волокон. В центре этих малых волокон миофибриллы отсутствуют или очень рыхло упакованы.
Гистохимические реакции выявляют в центральных областях высокую активность окислительных ферментов. Измененные мышечные волокна в центре очень напоминают эмбриональные мышечные клетки или миотубулы. В связи с этим заболевание получило название митубулярной миопатии. Другое ее название - центронуклеарная, что обусловлено скоплением в центральной части мышечного волокна группы ядер.
Клиника характеризуется генерализованной мышечной гипотонией, слабостью глазодвигательных мышц (двусторонний умеренный птоз, парез наружных мышц глаза), умеренными костными деформациями.
Течение заболевания часто стационарное, но может иметь место регресс симптомов или со слабым прогрессированием. В большинстве случаев клинические симптомы отмечаются с первых дней жизни, но иногда проявляются впервые у взрослых.
Миопатия с врожденной диспропорцией типов волокон
Мышечные волокна делятся на два типа: I тип - красные волокна, II тип - белые. В последнее время II тип подразделяют на II A и II Б типы. В норме типы I и II имеют одинаковые размеры, их соотношение неодинаково в различных мышечных группах, но это различие довольно постоянно. При изучении врожденных форм миопатий с клинической картиной "floppy baby", было показано, что у некоторых больных патоморфологическое исследование выявляет единственный дефект - уменьшение в размерах типа I мышечных волокон и их преобладание, и сохранность величины увеличения в размере типа II волокон.
Иногда отмечается гипертрофия II A типа и отсутствие II Б типа волокон. Каких-либо других специфических изменений не выявляется. Клиническая картина характеризуется генерализованной слабостью, гипотонией, включающей дыхательную мускулатуру. В половине случаев отмечаются мышечные контрактуры разной степени выраженности.
Термин "врожденная миопатия с диспропорцией типов волокон" используется для описания новорожденных с гипотонией и иногда с артрогрипозом.
Немалиновая (нитеобразная) миопатия - наиболее частая форма. Патология состоит в наличии нитеобразных структур вдоль всего длинника пораженных мышечных клеток. Заболевание передается как по аутосомно-рециссивному, так и по аутосомно-доминантному типу. В основном семейные случаи заболевания, но все чаще спорадические случаи.
Клиника проявляется мышечной слабостью диффузного типа, включая мимические мышцы, которая отмечается с рождения. Слабость может распространяться на дыхательную мускулатуру.
Тяжесть состояния широко варьирует - от очень легкой до выраженной слабости с гипотрофией, приводящей к инвалидности. Больные отстают в двигательном развитии, позже начинают сидеть, ходить. Характерно сочетание мышечных симптомов с диспластическими изменениями скелета в виде вытянутой формы лицевого черепа, ретрогнатии «готического неба», деформации грудной клетки, позвоночника.
При биохимическом исследовании находят некоторое повышение активности креатинфосфокиназы в сыворотке крови.
Митохондриальные миопатии - группа миопатий, в которых специфическим и единственным морфологическим нарушением является патология митохондрий. Наследуются по аутосомно-рециссивному типу. Эта группа миопатий является гетерогенной, поскольку она представлена различными типами нарушения митохондрий.
При митохондриальных миопатиях морфологически может иметь место увеличение числа митохондрий (плеокониальная миопатия), увеличение их размеров (мегакониальная миопатия), неправильное расположение крист, наличие различных включений.
Плеокониальная миопатия - характеризуется эпизодическими приступами, начинающимися с жажды, рвоты, выраженной потребности в соли, профузного пота. Развивается мышечная слабость вплоть до вялой тетраплегии с отсутствием сухожильных рефлексов. Нарушается также речь и глотание. Приступ длится 10-14 дней. Выраженная мышечная слабость наблюдается с рождения, с возрастом может даже несколько уменьшаться. При морфологическом исследовании в мышечных волокнах обнаружены митохондрии гигантских размеров.
Илеокониальная миопатия - характеризуется медленно прогрессирующей слабостью и похуданием мышц проксимальных отделов конечностей. Отличительной особенностью заболевания явилось наличие приступов усиления мышечной слабости до состояния параличей. Такие приступы длились каждый раз от 10 до 14 дней, при этом появлялась потребность в приеме большого количества поваренной соли. При морфологическом исследовании биоптата мышц было обнаружено огромное количество митохондрий средней величины. Скопления митохондрий замещали миофибриллярный аппарат и располагались в околоядерных областях.
Врожденная мышечная дистрофия - гипотония и артрогрипоз определяются с рождения. Сухожильные рефлексы либо сохранны, либо отсутствуют, но из-за контрактур суставов их бывает трудно оценить. Врожденные контрактуры могут вовлекать различные суставы, но наиболее часто они приводят к кривошее и косолапости. Характерный симптом - врожденный вывих бедра.
Лабораторные исследования: возможно повышение уровня КФК в сыворотке крови.
Инструментальные исследования:
1. Компьютерная томография головного мозга для исключения органического поражения.
2. Электроэнцефалография (ЭЭГ) - изменения ЭЭГ неспецифичны.
3. Электромиография (ЭМГ) - выявляет первичный мышечный дефект, снижение амплитуды ЭМГ максимального напряжения, длительность потенциалов ДЕ уменьшена, увеличение числа полифазных потенциалов без увеличения их длительности, иногда ЭМГ имеет смешанный тип.
4. Магниторезонансная томография (МРТ) головного мозга - в настоящее время является методом выбора при диагностике пороков и аномалий и дегенеративных изменений головного мозга.
5. Нейросонография - выявляет наличие структурных изменений, расширение желудочковой системы, пороки и дизгенезии до закрытия родничка.
6. ЭКГ - изменения, указывающие на обменно-дистрофические изменения в миокарде.
Показания для консультаций специалистов:
1. Логопед - для выявления нарушения речи, дизартрий.
2. Психолог - для определения психического статуса ребенка.
3. Ортопед - для выявления контрактур, костных деформаций.
4. Протезист - для подбора ортопедической обуви, фиксирующих лонгет.
5. Окулист - осмотр глазного дна, выявление страбизма, нарушений зрения.
6. Кардиолог - выявление обменно-дистрофических и диспластических изменений сердца.
7. ЛОР-сурдолог - для определения тугоухости.
Минимум обследования при направлении в стационар:
1. Общий анализ крови.
2. Общий анализ мочи.
3. Кал на яйца глист.
Основные диагностические мероприятия:
10. Электромиография (ЭМГ).
14. Определение креатинкиназы в сыворотке крови.
Перечень дополнительных диагностических мероприятий:
1. Компьютерная томография головного мозга.
5. МРТ головного мозга.
7. R-графия грудной клетки.
8. УЗИ органов брюшной полости.
10. ЛОР сурдолог.
14. Мышечная биопсия.
Дифференциальный диагноз
Заболевания
Начало заболевания
ЭМГ
Течение заболевания
С рождения или в раннем детском возрасте, проявляется снижением мышечной силы и гипотонией мышц.
Снижение амплитуды ЭМГ максимального напряжения, длительность потенциалов ДЕ уменьшена, увеличение числа полифазных потенциалов.
Доброкачественное течение, стационарное или медленно прогрессирующее. При некоторых формах двигательные функции с возрастом улучшаются.
ДЦП. Атонически-астатическая форма
С рождения, на первый план выступает выраженная мышечная гипотония.
При произвольном сокращении регистрируется интерференционная кривая со сниженной амплитудой; фибрилляции, фасцикуляции отсутствуют.
Не прогрессирует, по мере роста и развития ребенка происходит регресс некоторых симптомов.
Заболевание начинается на 1-ом году жизни или обнаруживается уже при рождении. Спонтанная двигательная активность резко ослаблена.
Дегенерация мотонейронов спинного мозга, проявляющаяся ритмом частокола, II тип. В тяжелых случаях полное биоэлектрическое молчание.
Неуклонно прогрессирующее заболевание, обусловленное уменьшением и дегенерацией клеток мотонейронов спинного мозга.
Лечение
Тактика лечения
Цель лечения: улучшение трофики мышц и проведение импульса по нервному волокну и через мионевральный синапс; улучшение двигательной активности, социальная адаптация, профилактика патологических поз и деформаций.
Немедикаментозное лечение:
- массаж общий, тонизирующий;
- занятия с логопедом, психологом.
Медикаментозное лечение:
1. Нейропротекторы: церебролизин, актовегин, пирацетам, пиритинол, гинкго-билоба.
2. Стимулирующая терапия: прозерин, дибазол, галантамин, оксазил, нейромидин.
3. Ангиопротекторы: винпоцетин, циннаризин.
4. Витаминотерапия: витамины группы В - тиамин бромид, пиридоксин гидрохлорид, цианокобаламин, аевит, фолиевая кислота, токоферол, ретинол, эргокальциферрол.
5. Комплексы аминокислот: метионин, глицин.
6. Анаболические стероиды: неробол, ретаболил, оротат калия.
Профилактика осложнений:
- предупреждение контрактур, патологических поз;
- профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение:
- регулярные занятия ЛФК;
- обучение родителей навыкам массажа, ЛФК;
- носить ортопедическую обувь.
Перечень основных медикаментов:
1. Аевит, капсулы
2. Актовегин ампулы по 80 мг, 2 мл
3. Нейромультивит (витамины В1, В6, В12), таблетки
4. Оротат калия, таблетки
5. Пирацетам, ампулы по 5 мл, 20%
6. Пиридоксин гидрохлорид, ампулы по 1 мл, 5%
7. Прозерин, ампулы 0,05%, 1 мл
8. Прозерин, таблетки 0,015
9. Тиамин хлорид ампулы, 1 мл, 5%
10. Фолиевая кислота, таблетки 0,001
11. Цианокобаламин, ампулы 200 и 500 мкг
Дополнительные медикаменты:
1. Винпоцетин, таблетки 5 мг
2. Галантамин, ампулы 0,25%, 1мл
3. Гинкго-Билоба, (танакан) таблетки 40 мг
4. Глицин, таблетки 0,1
5. Дибазол, таблетки 0,02
6. Колекальциферол 1 мл, 15 тыс. МЕ
7. Луцетам, таблетки 0,4
8. Магне В6, таблетки
9. Нандролона деканоат (ретаболил), ампулы 1 мл, 50 мг
10. Неуробекс, таблетки
11. Оксазил, таблетки 0,001
12. Пирацетам, таблетки 0,2
13. Пиритинол, суспензия или таблетки 0,1
14. Токоферолола ацетат (витамин Е), капсулы 200 МЕ
15. Церебролизин, ампулы 1 мл
16. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
1. Улучшение двигательной активности.
2. Улучшение мышечного тонуса.
3. Повышение силы мышц.
4. Приобретение навыков самообслуживания.
5. Улучшение эмоционального и психического тонуса ребенка.
Госпитализация
Показания к госпитализации (плановая): двигательные расстройства, мышечная слабость, ослабление или отсутствие сухожильных рефлексов, нарушение движений, походки, костные деформации.
Лучевая диагностика миодистрофий
Методы лучевой визуализации на сегодняшний день наиболее эффективны в диагностике заболеваний, характеризующихся дистрофическими изменениями мышечной ткани.
Они позволяют обнаружить патологические изменения на самой ранней стадии и используются в ходе дальнейшей терапии для контроля результатов лечения.
Особая роль отводится этим методам в раннем выявлении и своевременном лечении пациентов с наследственными скелетно-мышечными заболеваниями.
Методы визуализации, благодаря своей безопасности и высокой результативности, могут использоваться для выявления различных миодистрофий и миопатий не только у взрослых, но и у детей.
И если первые дифференциально-диагностические подходы в МРТ были сосредоточены главным образом на визуализации поражения определенной группы мышц нижних конечностей, современные исследования позволяют оценить состояние различных отделов скелета, мышцы туловища, плечевого пояса и верхних конечностей.
Скелетно-мышечная лучевая диагностика в целом и магнитно-резонансная томография (МРТ), в частности, все чаще используются для характеристики степени поражения мышечной ткани и помогает выявить различия между дистрофическими и недистрофическими заболеваниями. А стандартизованные классификации степени поражения мышц при проведении МРТ позволят оперативно оценить патологический процесс в каждой мышце.
Специфическая картина изменений при скелетно-мышечной МРТ может быть полезной для дифференциации отдельных форм поясно-конечностных миодистрофий(ПКМД) и дистрофинопатий.
Компьютерная томография (КТ) позволяет оценить глубокие мышечные структуры.
А ультразвуковое исследование(УЗИ), в свою очередь, провести динамические исследования сокращающихся мышц на предмет патологической мышечной активности. Благодаря отсутствию лучевой нагрузки, проводить исследование можно детям любого возраста.
В дополнение к неврологическому обследованию и нейрофизиологической оценке, скелетно-мышечная визуализация становится все более важным диагностическим инструментом для оценки мышечной ткани, показывающим степень и характер поражения.
Поясно-конечностные мышечные дистрофии (ПКМД) и дистрофинопатии имеют свои характерные особенности, которые важно учитывать при дифференциальной диагностике той или иной патологии.
Скелетно-мышечные изображения в этом случае играют важную роль в подтверждении клинического диагноза, сужая широкую гамму возможных заболеваний и облегчая задачи инвазивной диагностики, такой как биопсия мышц.
С помощью инновационных направлений в лучевой диагностике стало возможным выявлять большое количество наследственных, метаболических и воспалительных заболеваний мышц и, таким образом, предотвратить их дальнейшее развитие.
При диагностике специалисту следует учитывать ключевые моменты клинической картины - наиболее распространенные формы поясно-конечностных мышечных дистрофий, дистрофинопатий и их МР-семиотику, а также МРТ - алгоритм дифференциальной диагностики миодистрофий.
Методы визуализации
Поперечно-полосатая мускулатура является активно развивающейся тканью и находится под влиянием многих факторов, таких как физическая нагрузка, возраст и пол и.т.д. [5]. Возраст является одним из наиболее важных аспектов, влияющих на строение мышечной ткани [6]. В детстве по мере роста ребенка толщина мышц быстро растет. После полового созревания, в мышцах появляются гендерные различия (у мужчин после периода полового созревания объем мышечной ткани больше, чем у женщин). Пик объема мышечной ткани приходится на возраст 25-40 лет, с последующим снижением. Влияние возраста и пола на формирование поперечно-полостатой мускулатуры, однако, не является линейным и отличается для каждой группы мышц 6.
С помощью методов лучевой диагностики можно визуализировать физиологические изменения мышц и дифференцировать их от патологических процессов.
Большой интерес возможности диагностической визуализации также представляют для мониторинга лечения метаболических скелетно-мышечных заболеваний, и первые полученные результаты обнадеживают [40, 41]. Потенциальная роль новейших МР-техник, таких как перфузионно-взвешенные изображения, диффузионно-взвешенные изображения и МР-спектроскопия, а также использование контрастных препаратов в настоящее время не определена. Можно предположить, что некоторые из этих методов смогут увеличить точность обнаружения тонких изменений в поперечно-полосатых мышцах и будут способствовать более глубокому пониманию основных патофизиологических механизмов 35.
Первые дифференциально-диагностические подходы в МРТ
Первоначально, при подозрении на миодистрофию использовались МР-протоколы для отдельных анатомических областей, например, таких как нижние конечности и область таза. Впоследствии на высокопольных МР-томографах стало возможным осуществлять сканирование всего тела за 30-40минут, с одновременной оценкой практически всех групп поперечно-полосатых мышц, что облегчает постановку правильного диагноза. Кроме того, МРТ всего тела помимо мышц позволяет верифицировать патологию других тканей и органов, которые могут быть затронуты у пациентов с наследственными нервно-мышечными заболеваниями - это паренхиматозные органы брюшной полости, пищевод и сердце. [28, 29].
Хотя первые дифференциально-диагностические подходы в МРТ были сосредоточены главным образом на визуализации поражения определенной группы мышц нижних конечностей, настоящие исследования направлены на использовании информации от различных отделов скелета, включая МРТ мышц туловища, плечевого пояса и верхних конечностей, чему, безусловно, способствует появление современных быстрых МР-последовательностей и приложений. Высокопольные МРТ все чаще используются в клинических условиях [36, 37]. Из-за значительного увеличения отношения сигнал-шум, in vivo удается получать изображения с высоким пространственным разрешением за короткое время [27, 38, 39].
Диагностическая ценность МРТ с контрастным усилением при миодистрофиях на сегодняшней день остается сомнительной. Отсроченные контрастные серии в ряде случаев могут быть полезны для выявления гиперфиксации препаратов гадолиния соединительной тканью. Проведенные опыты на животных с контрастными препаратами, тропными к поврежденной поперечно-полосатой мускулатуре, показали обнадеживающие результаты не только для дифференциации «здоровых» и «больных» мышц, но и для более глубокого понимания патогенеза мышечного повреждения при дистрофических расстройствах [30, 31].
Количественные МРТ методы, такие как Т2-картирование, количественный подсчет мышечного жира, магнитно-резонансная спектроскопия и МР-перфузия потенциально могут быть полезны для анализа степени патологических изменений в поперечно-полосатых мышцах 32. В частности, измерение скорости мышечного кровотока помогает разграничить воспалительный (увеличение перфузии микроциркуляторного русла) и дегенеративный/дистрофический характер поражения.
Дальнейшие исследования позволят определить место данных методик в алгоритме обследования больных миодистрофиями.
Магнитно-резонансная томография (МРТ)
МРТ все чаще используется в обследовании пациентов с подозреваемой или доказанной врожденной или метаболической нервно-мышечной дистрофией. МРТ обеспечивает высокий контраст мягких тканей, что позволяет провести отличную оценку поперечно-полосатых мышц, их формы, объема (гипотрофия, гипертрофия), а также структуры [1, 2]. Из-за отсутствия ионизирующего излучения, МРТ стала ценным методом визуализации у детей, даже при необходимости седации [3].
Протокол МРТ для скелетно-мышечной визуализации включает несколько последовательностей, таких как T1-взвешенные (Т1-ВИ) и Т2-взвешенные (Т2-ВИ) турбо спино-эхо последовательности, а также Т2 взвешенные последовательности с использованием режимов жироподавления - по типу STIR или спектрального подавления жировой ткани (Т2 FS) . Основной плоскостью для получения изображений является аксиальная, достаточная толщина среза обычно составляет 5-7мм. В случае необходимости выполняются дополнительные исследования в сагиттальной и корональной плоскости или перпендикулярно ходу мышечных волокон.
Дистрофические изменения, такие как жировая дегенерация мышечных волокон может быть легко и точно обнаружены с применением T1-ВИ и T2-ВИ последовательностей (рисунок 2).
Рисунок 2. Представлены МР-томограммы бедер и голеней мужчины 34 лет с ПКМД 2А, выполненных в аксиальной плоскости в режиме Т1-ВИ
Отмечается асимметричная билатеральная жировая дегенерация отдельных мышц бедер и голеней (указаны белыми и черными стрелками):
- полное жировое замещение мышечной ткани полуперепончатых мышц и длинных головок бицепса на бедре, камбаловидных мышц на голенях - тотальная жировая дегенерация (4 стадия по шкалам Mercuri и Fischer, 3 стадия по шкале Kornblum);
- головки четырехглавых мышц, медиальные и латеральные головки икроножных мышц имеют неровные нечеткие контуры, узурпированы по типу ткани, «изъеденной молью», с множественными разнокалиберными участками повышенного сигнала, занимающими от 20% до 80% их объема - полиморфная картина с поражением мышц от начального до выраженного (1-3 стадия по шкалам Mercuri и Fischer, 1-2 стадия по шкале Kornblum)
Кроме того, воспалительные изменения, такие как отек мышечной ткани также хорошо визуализируются при МРТ в T2 FS последовательности.
В свою очередь, с помощью техник жироподавления МРТ позволяет обнаружить мельчайшие участки отека мышечной ткани (токсического, метаболического или воспалительного характера). Зоны миофибриллярного отека свидетельствует об активности процесса и могут быть мишенью при необходимости биопсии (рисунок 3).
Рисунок 3. На T2 FS в аксиальной плоскости у пациента 15 лет с ПКМД 2B (дисферлинопатия) определяется повышение МР-сигнала (указаны стрелками) от головок четырехглавых мышц бедра (латеральной, медиальной и промежуточной широких мышц), а также приводящих мышц (преимущественно поражены большие приводящие мышцы), характеризующей ее воспалительный отек и рабдомиолиз на ранних стадиях миодистрофии. Рядом авторов было убедительно показано, что МРТ обладает более высокой чувствительностью в выявлении дистрофических изменений по сравнению с КТ [22, 23]. Степень мышечной дистрофии определяется при помощи оценочных шкал 25. Большинство разработанных оценочных шкал основаны на степени жировой перестройки мышечной ткани, начиная с отсутствия жира в мышце и заканчивая тотальной липодистрофией.
Стандартизованные классификации степени поражения мышц
Стандартизованные классификации степени поражения мышц позволяют быстро и достоверно оценить характер и стадию вовлечения каждой мышцы. Таблица МРТ классификации, применяемые для визуальной оценки дистрофических изменений поперечно-полосатой мускулатуры
Компьютерная томография (КТ)
Еще недавно КТ широко использовалась у пациентов с наследственными нервно-мышечными расстройствами [2, 22-24]. КТ - быстрый метод визуализации, который легко выполним и стандартизован, позволяет оценить структуру и форму мышц, а также дистрофические изменения (в частности, жировое перерождение). С помощью КТ можно оценить глубокие мышечные группы, а новые мультидетекторные КТ-сканеры обеспечивают высокое пространственное разрешение с возможностью мультипланарных и 3D реконструкций. К сожалению, одним из наиболее важных недостатков КТ по сравнению с УЗИ и МРТ, ведущим к почти полной замене метода, является относительно высокая доза облучения, что делает применение КТ небезопасным, особенно у детей. По этой же причине, исследование всего тела для визуализации пораженных групп мышц нежелательны. Еще одним недостатком КТ является невысокая мягкотканая контрастность, что существенно снижает чувствительность в выявлении воспалительных изменений (например, отеков мышечной ткани), которые могут быть предикторами формирующихся миопатий и миодистрофий.
Ультразвуковое исследование (УЗИ)
УЗИ - широко распространенный и эффективный метод в оценке пациентов с подозрением на патологию мышечной ткани [10]. УЗИ является относительно дешевым и легкого выполнимым методом, позволяющим визуализировать поперечно-полосатые мышцы с высоким пространственным разрешением (> 0,1 мм). Основным преимуществом УЗИ является отсутствие лучевой нагрузки, что делает его незаменимым инструментом скрининга миопатий у детей (чувствительность колеблется в диапазоне от 25% для выявления недистрофических миопатий до 100% при дистрофических миопатиях) [10, 19, 20]. УЗИ также позволяет проводить динамические исследования сокращающихся мышц на предмет патологической мышечной активности, например фасцикуляций.13. К основным недостаткам метода следует отнести ограниченную эффективность в оценке глубоких групп мышц по сравнению с поверхностными, причинами которой служат как физические аспекты получения изображения (отражение и поглощение звуковой волны), так и особенности анатомии и расположения мышц. Необходимо также помнить о высокой оператор-зависимости УЗИ и сложностей проведения стандартизованного исследования (например, в соответствии с определенными анатомическими ориентирами). [2, 14].
Тем не менее, УЗИ приложения широко и регулярно задействованы для морфологической характеристики мышц (атрофия, гипертрофия, изменения мышечной архитектоники - в том числе дистрофического характера (рисунок 1)).
Рисунок 1. Ультразвуковые изображения четырехглавой мышцы у здорового человека (а) и пациента с мышечной дистрофией Дюшенна (b).
У здорового человека мышцы в основном гипоэхогенны с наличием многочисленных соединительно-тканных перегородок (наружный и внутренний перимизий) (а).
У пациента с мышечной дистрофией определяется повышение эхогенности мышцы с ее мелкозернистой перестройкой из-за замены нормальных мышечных волокон соединительной и жировой тканью (b). European Radiology 2010 October; 20(10).
Для количественной оценки степени жировой дистрофии по УЗИ есть несколько классификаций (например, шкала Heckmatt), а также компьютерные алгоритмы количественного анализа эхосигнала. 19. Эффективной методикой верификации окончательного диагноза является биопсия мышечной ткани под УЗИ-контролем. [10, 21].
Поясно-конечностные мышечные дистрофии (ПКМД) и дистрофинопатии
ПКМД - группа прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового пояса, конечностей. Распространенность ПКМД широко варьирует в различных популяциях и составляет от 5 до 70 больных на 1 миллион населения. Типичными клиническими проявлениями ПКМД являются нарушения походки (переваливающаяся или «утиная» походка), «осиная» талия, приемы Говерса (подъем лесенкой из положения на корточках), гиперлордоз в поясничном отделе позвоночника, «крыловидные» лопатки, симптом «дряблых надплечий», сухожильная гипорефлексия, мышечная гипотония и гипотрофия.
На данный момент выделено 13 нозологических форм ПКМД с аутосомно-рецессивным (II тип) наследованием и 7 с аутосомно-доминантным (I тип). Среди аутосомно-рецессивных форм наиболее распространены ПКМД 2A (от 30 % в Бразилии до 80% среди басков Испании от всех выявленных случаев) и ПКМД 2B (от 20% до 40% всех случаев). На группу саркогликанопатий (ПКМД 2C-2F) приходится еще около 20-25% всех случаев ПКМД (ПКМД 2C широко распространена в Тунисе; ПКМД 2D - в Европе, США и Бразилии, а ПКМД 2E и ПКМД 2F - в Бразилии). ПКМД 2I нередко встречается у жителей Северной Европы. Остальные формы ПКМД встречаются редко, и в основном обнаруживаются в изолированных группах населения.
Дистрофинопатии — это рецессивные заболевания, обусловленные мутацией в 21 локусе короткого плеча Х-хромосомы, в крупном гене, который кодирует белок дистрофин (филаментный белок, который выполняет структурные и «якорные» функции). Выделяют две формы дистрофинопатии - более тяжелую мышечную дистрофию Дюшенна и более благоприятную мышечную дистрофию Беккера.
Поясно-конечностные дистрофинопатии и МР-семиотика
Дебют в возрасте от 10 до 30 лет. Раннее
вовлечение в процесс икроножных
мышц, что проявляется затруднениями
ходьбы на пятках. Показано, что
заболевание может встречаться в двух
клинических вариантах, наблюдаемых у
членов одной и той же семьи -
поясно-конечностная мышечная
дистрофия и дистальная
миопатия Миоши.
Выраженные дистрофические
изменения мышц передней и
задней поверхности бедра за
исключением портняжной и
нежной мышц.
Диагностика врожденной мышечной дистрофии на МРТ, КТ

Обзор
МРТ В МИОЛОГИИ
Нервно-мышечные болезни — группа гетерогенных болезней с различными клиническими проявлениями, главными из которых являются прогрессирующая мышечная слабость и утомляемость, различный возраст дебюта, повышенный уровень фермента креатинфосфокиназы (КФК) и дистрофические изменения с некрозами и регенерацией мышечных волокон. НМЗ являются одними из самых распространенных среди наследственных болезней человека. К сегодняшнему дню разработаны диагностические критерии основных форм наследственных НМЗ, изучены отдельные звенья их патогенеза. Однако выраженная клинико-генетическая гетерогенность НМЗ обусловливает значительную сложность для их дифференциальной диагностики. При установлении диагноза учитываются возраст появления первых клинических симптомов заболевания, тип миодистрофического поражения (дистальный и/или проксимальный), локализация атрофий, наличие или отсутствие псевдогипертрофий, фасцикуляций, расстройств чувствительности, крампи, скелетных деформаций, суставно-мышечных контрактур, состояния мышечного тонуса и сухожильных рефлексов, а также темп течения и прогрессирования заболевания. Большое значение имеют анализ родословной пробанда, а также типы наследования, которые при НМЗ могут быть аутосомно-доминантным, аутосомно-ре- цессивным, Х-сцепленным, могут встречатся и спорадические случаи (мутации de novo). Диагностически значимыми являются следующие методы: биохимический анализ крови, электронейромиография — ЭНМГ (стимуляционная и игольчатая), ультразвуковое исследование мышц, магнитная резонансная томография мышц (МРТ мышц), морфологическое исследование биоптата мышечной ткани, молекулярно-генетические исследования. В биохимическом анализе крови исследуется активность КФК, лактатдегидрогеназы (ЛДГ). Повышение активности КФК свидетельствует об активном распаде мышечных волокон (рабдомиолиз). Показатель активности КФК весьма вариабелен, так как активность КФК при врожденных миопати- ях обычно варьирует в пределах физиологической нормы или может быть повышена в 5-10 раз, а при прогрессирующих мышечных дистрофиях и пояс- но-конечностных мышечных дистрофиях возможно ее повышение в сотни раз. Однако повышение КФК не всегда означает наличие НМЗ, мы отмечали повышение содержания КФК у некоторых пациентов с детским церебральным параличом, поствакцинальными осложнениями полиомиелита, анафилаксией. Активность ЛДГ, как правило, варьирует в пределах физиологической нормы или может быть несколько повышена (до 2-3 раз). Магнитно-резонансная томография при первично-мышечной патологии используется в клинической практике более 20 лет. Широкое внедрение МРТ в первые годы было ограничено чрезмерной продолжительностью исследования и невозможностью обследования более одного сегмента. Около 12 лет назад в клиническую практику была внедрена МРТ всего тела,ставшая полезной диагностической методикой в ходе определения типа заболевания. Данный метод дает возможность получить объективную информацию о дегенеративных изменениях, жировой инфильтрации, выявить наличие отека и воспалительных изменений скелетной мускулатуры. Эта методика также достаточно четко позволяет разграничить анатомические структуры исследуемой области, можно выделить отдельные мышцы, сосудисто-нервные пучки, подкожно-жировую клетчатку, фасции, кости.
Показания для МРТ скелетных мышц
Развитие очевидной мышечной симптоматики, но при этом не получено объективных данных за мышечную патологию (особенно при неубедительных или неспецифических результатах мышечной биопсии), или когда данные обследования вызывают сомнение. • Исследование направлено на выбор мышцы, которую потенциально можно использовать для повторной биопсии; • Наличие мышечной патологии, симптомы которой предполагают узкий диагностический поиск. • Подозрение на миопатию вследствие специфической клинической картины, но без обнаружения первично-мышечного паттерна при ЭМГ. • Диагностировано (предполагается) редко встречающаяся патология мышц. МРТ мышц проводится с целью уточнения паттерна поражения мышц всего тела; • Диагностировано мышечное заболевание, для которого необходимо определить паттерн распределения и степень поражения мышц; • Все ситуации, в которых после мультидисциплинарного обсуждения диагноз попрежнему остается неясным. МРТ мышц может расцениваться как информативная методика для постановки диагноза. Большинство современных томографов позволяют осуществлять исследование всего тела. Для старых моделей данное исследование возможно при наличии телесной катушки, интегрированной в аппарат. Современные МРТ-аппараты снабжены несколькими катушками, соединенными друг с другом (для головы / шеи, грудной клетки, брюшной полости и ног). Система должна позволять покрывать голову, шею, туловище, а также конечности. Большая часть исследований всего тела в онкологии игнорирует верхние конечности, что недопустимо при проведении МРТ мышц, где плечи, предплечья и кисти должны быть включены в исследование. В связи с этим поперечное поле не должно быть слишком маленьким. Поля от 50 до 55 см хватает для большинства взрослых пациентов. Для педиатрического контингента, в частности детей ростом ниже 120 см, все аппараты позволяют проводить удовлетворительное обследование. Помимо оборудования, для проведения адекватного исследования радиологическая команда должна иметь опыт работы с пациентами-инвалидами, как взрослыми, так и детьми. Для пациентов, страдающих дыхательной недостаточностью, МРТ-исследование должно проводиться исключительно в устройствах, имеющих вентиляционные приборы, и при соблюдении условий амагнетизма, а также при наличии доступной реанимационной и анестезиологической помощи, что позволяет транспортировать и курировать сложных пациентов во время исследования
Диагностика мышечной дистрофии Дюшенна

Как диагностировать мышечную дистрофию Дюшен: Мышечная дистрофия Дюшенна— это наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первичная диагностика мышечной дистрофии Дюшенна потребует проведения генетических анализов и последующей консультации у невролога. В качестве дополнительного обследования врач может назначить МРТ позвоночника на аппарате 3 Тесла и электромиография.
Какой врач лечит мышечную дистрофию Дюшенна: При симптомах мышечной дистрофии Дюшенна следует сначала обратиться к врачу неврологу.
Симптомы мышечной дистрофии
Основным признаком мышечной дистрофии является прогрессирующая мышечная слабость. Конкретные симптомы начинают развиваться в разном возрасте и в разных группах мышц, в зависимости от типа мышечной дистрофии.
Мышечная дистрофия типа Дюшенна
Это наиболее распространенная форма, которая чаще встречается у мальчиков. Симптомы, которые обычно проявляются в раннем детстве:
- частые падения
- трудности с подъемом из положения лежа или сидя
- проблемы с бегом и прыжками
- измененная походка, ходьба на пальцах ног
- большие икроножные мышцы
- мышечная боль и скованность
- задержка роста и развития.
Мышечная дистрофия Беккера
Симптомы похожи на признаки мышечной дистрофии Дюшенна, но, как правило, мягче и прогрессируют медленнее. Симптомы обычно начинаются в подростковом возрасте, но могут возникнуть и после 20 лет.
Другие виды мышечной дистрофии
Некоторые виды мышечной дистрофии определяются конкретной особенностью или областью локализации симптомов в организме. Примеры:
- миотонический синдром. Состояние характеризуется неспособностью расслабить мышцы после сокращений. Мышцы лица и шеи, как правило, страдают первыми. Пациенты с такой формой, как правило, имеют длинные, тонкие черты лица, опущенные веки и ненормально длинные шеи
- фациоскапулохумерал. Мышечная слабость обычно развивается в лице, бедре и плечах. Ключицы могут сильно выпирать. Заболевание начинается в подростковом возрасте, но также может начаться в детстве или в возрасте 50 лет
- врожденный синдром атрофии мышц. Этот тип проявляется при рождении или до 2 лет. Некоторые формы прогрессируют медленно и вызывают только легкую инвалидность, в то время как другие вызывают необратимые нарушения
- дистрофия конечностей. Мышцы бедра и плеча обычно затрагиваются в первую очередь. Пациенты с этим типом мышечной дистрофии могут испытывать трудности с подъемом передней части стопы и поэтому могут часто спотыкаться. Болезнь развивается в детстве или подростковом возрасте.
Следует обратиться за консультацией к неврологу при выявлении признаков мышечной слабости, таких как повышенная неуклюжесть и частые падения.
Причины мышечной дистрофии
Определенные гены участвуют в создании белков, которые формируют мышечные волокна. Мышечная дистрофия возникает в случае мутации одного из функционирующих генов.
Каждая форма мышечной дистрофии вызвана генетической мутацией, характерной для конкретного типа заболевания. Большинство этих мутаций наследуется.
Факторы риска
Пациенты с семейным анамнезом мышечной дистрофии подвергаются более высокому риску развития заболевания или передачи его своим детям.
Осложнения
Осложнения прогрессирующей мышечной дистрофии:
проблемы с ходьбой. Некоторым пациентам с мышечной дистрофией в конечном итоге нужно использовать инвалидную коляску
сокращение мышц или сухожилий вокруг суставов (контрактуры). Контрактуры могут еще больше ограничить мобильность
проблемы с дыханием. Прогрессирующая слабость может повлиять на мышцы, связанные с дыханием. Пациентам с мышечной дистрофией в конечном итоге может потребоваться устройство для дыхания, сначала ночью, но, возможно, и днем
сколиоз. Ослабленные мышцы могут быть не в состоянии удерживать позвоночник прямо
проблемы с сердцем. Мышечная дистрофия может снизить эффективность сердечной мышцы
проблемы с глотанием. Если затронуты мышцы, связанные с глотанием, могут развиться проблемы с питанием и аспирационная пневмония.
Диагностика мышечной дистрофии - обследования и тесты
Пациенту назначается медицинский осмотр и ряд специализированных обследований:
- исследование ферментов. Поврежденные мышцы выделяют в кровь ферменты, такие как креатинкиназа. Если у пациента не было травмы, но при этом высокий уровень креатинкиназы в крови, то предполагается заболевание мышц
- генетическое исследование. Образцы крови могут быть проверены на мутации в некоторых генах, которые вызывают типы мышечной дистрофии
- биопсия мышц. Небольшой образец мышечной ткани отбирается через разрез или с помощью полой иглы. Анализ образца ткани позволяет отличить мышечную дистрофию от других мышечных заболеваний
- мониторинг сердца (электрокардиография и УЗИ сердца). Данные методы используются для проверки функции сердца, особенно у пациентов с диагнозом миотоническая мышечная дистрофия
- мониторинг функции легких
- МРТ позвоночника
- электромиография. Для обследования тонкая игла-электрод вводится в мышечную ткань. Электрическая активность измеряется по мере расслабления и мягкого напряжения мышц. Изменения в структуре электрической активности могут подтвердить заболевание мышц.

Специализация: Педиатр
Где ведет прием: Клиника Дженерал Медикал Центр ( GMC) г. Санкт-Петербург

Статья
Применение магнитно-резонансной томографии в диагностике идиопатических воспалительных миопатий
Идиопатические воспалительные миопатии (ИВМ)- гетерогенная группа аутоиммунных заболеваний, общим признаком которых является воспаление поперечно-полосатой мускулатуры с развитием слабости, преимущественно проксимальных групп мышц конечностей. К ИВМ относятся дерматомиозит (ДМ) (в том числе ювенильный и паранеопластический), полимиозит (ПМ), спорадический миозит с включениями (СМВ) и иммуно-зависимая некротизирующая миопатия (ИЗНМ).
ИВМ - относятся к редким заболеваниям, так заболеваемость ДМ 1.4 на 100 000 населения, ПМ 3.8 на 100 000. СМВ наиболее часто встречающаяся миопатия после 50-ти лет. Его распространенность колеблется от 9.3 до 51.3 на миллион лиц после 50-ти лет.
Учитывая редкость ИВМ, разнообразие их клинических проявлений, наличие большого числа нервно-мышечных заболеваний с похожей симптоматикой, а также отсутствие простых диагностических тестов, подтверждающих диагноз, выявление их может представлять определенную сложность.
Длительное время диагноз ДМ и ПМ устанавливался на основании критериев Bohan и Peter 1975г. В 2017г были опубликованы классификационные критерии ИВМ, предложенные EULAR/ACR. Новые критерии впервые рассчитаны и на взрослых, и на детей, а также могут быть применены у больных без биопсии мышц. Критерии включают две части - клинические признаки и лабораторные и/или морфологические признаки, оцениваемые в баллах. (таблица 1)
Если биопсия мышц не проводилась, диагноз вероятной воспалительной миопатии устанавливается при общей сумме балов ≥ 5.5, если данные биопсии учитывались - при сумме балов ≥ 6.7. Диагноз определенной ИВМ устанавливается без биопсии при сумме балов ≥ 7.5, а с биопсией ≥ 8.7.
Возможность установить диагноз ИВМ без проведения биопсии можно отнести к преимуществам данных критериев, так как биопсия не всегда доступна. Она является инвазивным вмешательством, способным вызвать осложнения, особенно у больных, получающих ГК, и не всегда является информативной. Диагноз ИВМ достаточно прост в случае классического дебюта заболевания, типичного распределения мышечной слабости, наличия кожных изменений, характерных для дерматомиозита. Однако, при медленно-прогрессирующей мышечной слабости, отсутствии изменений кожи и других признаков системного заболевания клинических (конституциональных симптомов, полиартрита, интерстициального поражения легких или синдрома Рейно) и иммунологических, могут возникать диагностические ошибки. Наиболее сложным в клинической практике может быть дифференциальный диагноз между полимиозитом, спорадическим миозитом с включениями и заболеваниями группы поясно-конечностных мышечных дистрофий (ПКМД) с поздним дебютом. Так у пациентов с ИВМ и ПКМД отмечается прогрессирующая мышечная слабость, повышен уровень мышечных ферментов, миографическая картина характеризуется первично-мышечным уровнем поражения, а световая микроскопия может выявлять признаки мышечного воспаления. Маммен проанализировал, какие варианты невоспалительных миопатий наиболее часто могут имитировать ПМ. Наиболее часто за ПМ принимают дисферелинопатию, калпаинопатию и лице-лопаточно-плечевую мышечные дистрофии, т.к. при данных заболеваниях в мышечном биоптате можно выявить признаки воспаления. Также диагноз ПМ может быть ошибочно установлен при гликогенозах (болезнь МакАрдла, Помпе). СМВ также часто принимают за ПМ, что приводит к необоснованному назначению глюкокортикоидов этой категории больных. А гипотиреоидная и самолимитирующаяся статиновая миопатия могут быть неверно трактованы как ИЗНМ. У ревматолога также возникают сложности в дифференциальном диагнозе ПМ с другими нервно-мышечными заболеваниями, для которых характерен иной, не мышечный уровень поражения периферического нервно-мышечного аппарата (болезнь двигательного нейрона, нарушение нервно-мышечной передачи, или полинейропатии), для которых также типично развитие мышечной слабости и, изредка, может встречаться повышение уровня «мышечных» ферментов. В таких случаях осмотр невролога и комплексное электромиографическое (ЭМГ) исследование позволяет исключить ПМ. В тоже время, в редких случаях ПМ может иметь медленно-прогрессирующее течение, в связи с чем пациенту предполагают наследственный характер болезни и иммуносупрессивная терапия не назначается вовремя. Таким образом, врачи ревматологи и неврологи нуждаются в дополнительных методах обследования, позволяющий оценить состояние мышц. В клинической практике используются различные методы визуализации мышц, основным из которых является магнитно-резонансная томография.
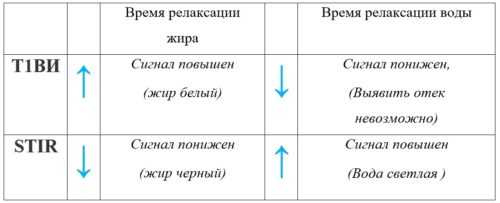
Более 20 лет при нервно-мышечных заболеваниях в клинической практике используется МРТ скелетных мышц. Данный метод дает объективную информацию о локализации и распространенности, симметричности патологического процесса, а также выявляет гипо- или гипертрофии различных мышц или мышечных групп. С помощью МРТ возможно более точно оценить избирательность поражения отдельных мышц в составе мышечных групп, которая не всегда выявляется при клиническом неврологическом обследовании. МРТ мышц — неинвазивный диагностический метод, позволяющий четко разграничить анатомические структуры исследуемой области: можно выделить отдельные мышцы, сосудисто-нервные пучки, подкожно-жировую клетчатку, фасции, кости. Кроме анатомических характеристик, МРТ позволяет оценить некоторые патофизиологические нарушения, в частности дегенеративные изменения, жировое замещение мышцы, наличие отека и воспалительных изменений скелетной мускулатуры. С помощью МР-томографа во фронтальной и аксиальной ориентации срезов и использованием Т1- и Т2-импульсных последовательностей мышц бедра и голени у больных с миопатиями получают серию изображений, позволяющий оценить состояние основных анатомических структур. Наиболее часто, в диагностике мышечных заболеваний используют аксиальную ориентацию срезов. Т1 взвешенное изображение позволяет оценить структуру мышц, их размер. Режим STIR (short tau inversion recovery) - приводит подавлению сигнала от жира, что позволяет отличить отек мышцы от жирового замещения. Применения режима STIR необходимо при диагностике ИВМ, т к позволяет визуализировать типичный для этой группы болезни воспалительный мышечный отек.
МР-последовательности и основные характеристики сред
При проведении МРТ основными анализируемыми параметрами являются мышечный отек (STIR), жировая дегенерация мышечной ткани и изменение размера мышц.
Мышечный отек - это неспецифический симптом, который может выявляться при ряде состояний: миозиты (аутоиммунный, инфекционный, саркоидный), рабдомиолиз, диабетический мионекроз, сосудистое повреждение (сосудистый инсульт), травматическое повреждение, после чрезмерной физической нагрузки, ранняя стадия оссифицирующего миозита, подострая денервация и радиационная травма.
Замещение мышц жировой тканью. Жировое замещение мышц может возникать как при первично-мышечных заболеваниях (наследственных мышечных дистрофиях), так и при хронических денервационных процессах (нейрональных заболеваниях, полинейропатиях), а также при длительной иммобилизации и на фоне приема глюкокортикоидов. Анализируется паттерн распределения данных изменений в мышцах у пациентов с миопатиями, что позволяет предположить нозологическую принадлежность мышечного заболевания и определить объем генетического исследования. Для определения степени дегенерации мышечной ткани при миопатиях используют шкалу E. Mercuri, в которой анализируются поперечные срезы мышц бедра и голени в режиме Т1.
Увеличение в размерах мышц, масс-эффект. Увеличение в размерах мышц может выявляться при пиомиозите, абсцессах, паразитарных заболеваниях, повреждениях (гематома), оссифицирующем миозите, мионекрозе, доброкачественных и злокачественных опухолях, фокальном пролиферативном миозите.
Преимуществом МРТ является большой объем анализируемых мышц, в отличие от мышечной биопсии, когда маленький по размеру биоптат берут из одной мышцы. В любом случае, при планировании выполнения биопсии мышц целесообразно провести МРТ, для более точного определения места взятия биоптата. При проведении биопсии важно, чтобы мышца имела признаки активного воспаления, но при этом с сохраненную мышечную структуру в Т1, т.к. при выраженным замещении мышцы жиром биопсия не информативна (что имеет значение в диагностике СМВ). Важно, что при подозрении на полимиозит, а МРТ следует направлять тех пациентов, у которых проведено ЭМГ исследование, и подтвержден первично-мышечный уровень поражения, т к при развитии денервации при МРТ может выявляться отек мышц, что может привести к неверной трактовке данного феномена и диагностическим ошибкам. Для оценки состояния мышц чаще всего проводится исследование мышц бедер и голеней, но имеется методика выполнения МРТ всего тела. Так, Huang и соавт. провели МРТ всего тела у 129 пациентов с ПМ и ДМ и у 105 пациентов выявили воспалительный отек мышц. Кроме этого были обнаружены 38 случаев интерстициального поражения легких, 12 опухолей (в том числе 5 злокачественных) и у больных 15 - остеонекроз костей. Таким образом, МРТ всего тела позволяет, наряду с отеком мышц, выявить ряд состояний, очень важных и актуальных для пациентов с ДМ и ПМ как в плане оценки состояния, так и терапии. В практическом отношении следует учитывать, что с целью дифференциального диагноза у пациентов с мышечными дистрофиями используется, как наиболее информативная, МРТ мышц бедер и голеней, поэтому в сложных дифференциально-диагностических случаях предпочтение следует отдать именно исследованию мышц нижних конечностей (бедра и голени). Интересно, что в отличие от наследственных миопатий, у больных ДМ/ПМ при исследовании в режиме Т1 в клинически слабых мышцах патологические изменения не выявляются (в течение первых лет от начала заболевания). Исключения составляют СМВ и некротизирующая миопатия, при которых параллельно с признаками воспаления, появляется жировая дегенерация мышцы.
Особенности МРТ в группе ИВМ: Важно подчеркнуть, что МРТ позволяет различить паттерны распределения поражения при разных клинических вариантах ИВМ. Так, при ДМ имеет место периферический отек мышц с паттерном по типу медовых сот, а также подкожный и фасциальный отек, тогда как для ПМ более характерны пятнистый или «туманный» паттерн распределения отека. При анализе МРТ картины мышц бедра у большой группы больных с различными клиническими вариантами ИВМ (101 с ИЗНМ, 176 ПМ, 219 ДМ, 17 пациентов с амиопатическим ДМ и 153 с СМВ) было показано, что в сравнении с пациентами с ДМ и ПМ у пациентов с ИЗНМ отмечается более выраженный мышечный отек, особенностью этих пациентов было раннее появление жирового замещения мышц и атрофии. Жировое замещение в первую очередь, затрагивало латеральный ротатор и ягодичные мышцы. Отек фасций наиболее характерен для пациентов с ДМ. Пациенты с ДМ и ПМ имели более сохранные мышцы бедер, по сравнению с пациентами с ИЗНМ.
Для СМВ типично сочетание как воспалительных изменений (которые выявляются при исследовании в режиме STIR), так и мышечной дегенерации с замещением жировой тканью мышечной. Для СМВ, как и для наследственных миопатий, типична избирательность поражения. Наиболее пораженными мышцами при СМВ четырехглавая мышца (за исключением относительно сохранной прямой мышцы бедра) и медиальная головка икроножной мышцы. Также для СМВ типичным является асимметричность поражения - на доминирующей стороне мышцы отличаются большей сохранностью. Tasca и соавт. выполнили МРТ мышц 17 пациентам с достоверным СМВ, 2 с вероятным СМВ и 118 пациентам с другими миопатиями. Были получены результаты, выявившие особенности поражения мышц при этом заболевании, свидетельствующие о высокой информативности данного метода в диагностике СМВ. Таким образом, МРТ мышц является надежным инструментом для пациентов с подозрением на СМВ, и м б особенно полезным у пациентов с ранней стадией заболевания, когда еще отсутствуют все типичные морфологические признаки этого заболевания. Продолжается сравнение информативности МРТ и биопсии мышц. В этом плане интересные данные получены норвежскими учеными, которые проанализировали 230 пациентов с ДМ и ПМ и выявили, что у 87% пациентов с полимиозитом и 82% пациентов с дерматомиозитом имеют место изменения по результатам МРТ, тогда как достоверные морфологические признаки получены у 70% пациентов с ПМ и 70% пациентов с ДМ. Для полимиозита и дерматомиозита типичным является повышение МР- сигнала в режиме STIR, что отражает наличие воспалительного отека в них. При ДМ признаки воспаления могут выявляться в коже и подкожно-жировой клетчатке (что отражает наличие панникулита в рамках ДМ). Учитывая высокую частоту выявления патологических изменений в мышцах при МРТ обсуждается возможность включения МРТ в диагностические критерии ИВМ. Недостатком данного метода исследования при ИВМ является отсутствие патогномоничных для ПМ и ДМ изменений. Для ВМ характерно наличие мышечного отека, который выглядит как усиление сигнала в режиме STIR, однако аналогичные изменения могут выявляться при денервационных процессах, повреждении, ишемии, мионекрозе и после чрезмерных физических нагрузок.
Клинический пример №1. Полимиозит. Пациентка Ф 73 лет: Впервые консультирована ревматологом в сентябре 2014г, когда стала отмечать небольшую мышечную слабость в мышцах бедер, тогда имели место жалобы на утомляемость и общую слабость. В течение 2014г года отмечала некоторое уменьшение мышечной силы, хотя при этом активно двигалась, могла поднять внука, но изредка чувствует неустойчивость при приседании на низкое и стало труднее вставать с низкого стула. В сентябре 2013г впервые в биохимическом анализе отмечались повышение АЛТ до 87 АСТ до 80 ед (КФК не исследовано), калий, СРБ - норма, Препараты, в том числе статины не принимает. В дальнейшем АЛТ и АСТ продолжало нарастать, уровень калия в норме, незначительная ТТГ 4,45 (норма до 4). 27.08.14 КФК 4340, миоглобин 849 (норма 73), АЛТ 232, АЛТ 158, ЛДГ креатинин 88, мочевина 4,8, о. белок 67г/л, СРБ в норме, калий 4,4, онкомаркеры в норме. Общий анализ мочи - норма, в повторном анализе 240, АСТ 153, КФК 3850, ЛДГ 1362. При осмотре в сентябре 2014: нормального питания. Кожные покровы чистые, мышечная сила 10 баллов во всех группах мышц. Чувствительных нарушений нет. В легких дыхание везикулярное, хрипов нет. Тогда мышечная слабость отсутствовала, пациентка была направлена к неврологу при и ЭМГ - признаки генерализованного активного первично-мышечного процесса, проведено МРТ мышц: практически норма, минимальный отек мышц в STIR. К лету 2015г пациентка не могла встать с корточек. С ноября 2015г - значительное прогрессирование мышечной слабости в мышцах плечевого пояса и бедер, туловища, стало трудно вставать с низкого стула, причесываться, снимать одежду через голову, (КФК 9500ед). Повторно МРТ выявило значительное повышение МР сигнала от мышц бедер и голеней. Проведен онкопоиск: гастроскопия, маммография, УЗИ полости малого таза, ранее КТ органов брюшной полости - без признаков ЗНО. При осмотре: Нормального питания, походка миопатическая, со стула встает с приемами Говерса, руки выше горизонтального уровня поднимает с затруднением. Мышечная сила снижена в дельтовидых и трехглавых мышцах до 6 баллов, в подвздошно-поясничных, четырехглавых мышцах до 7 баллов, в двуглавой мышце бедра до 8 баллов, при перемене положения из горизонтального в вертикальное положение - пользуется вспомогательными приемами.
Читайте также:
- Диагностика поражений сердца при тиреотоксикозе. Дифференциация поражений сердца при тиреотоксикозе
- Регуляция активности генов через питание
- Психогенные причины соматических заболеваний. Невроз органа
- Пункция абсцесса легкого. Введение антибиотика в полость абсцесса
- Ригидная молоткообразная деформация пальцев: атлас фотографий