Факоматозы - причины, симптомы, диагностика и лечение
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Туберозный склероз — генное заболевание, характеризующееся поражением нервной системы в виде эпилепсии и олигофрении, полиморфными кожными симптомами, опухолевыми и неопухолевыми процессами в соматических органах. Диагностический алгоритм состоит из обследования нервной системы (МРТ, КТ головного мозга, ЭЭГ), офтальмологического исследования, обследования внутренних органов (УЗИ, МРТ сердца, КТ почек, рентгенография легких, ректороманоскопия). Главными направлениями лечения являются: противоэпилептическая терапия, нейропсихологическая коррекция, наблюдение и своевременное хирургическое лечение новообразований.
МКБ-10
Общие сведения
Туберозный склероз (ТС) — наследственная нейроэктодермальная патология, проявляющаяся изменениями кожи, эпилептическими приступами, олигофренией (умственной отсталостью) и возникновением новообразований различной локализации. Наряду с нейрофиброматозом, болезнью Гиппеля-Линдау, синдромом Луи-Бар, болезнью Стерджа-Вебера и др., ТС относится к факоматозам. Заболеваемость составляет 1 случай на 30 тыс. населения, среди новорожденных — 1 случай на 6-10 тыс. Известны не только семейные, но и спорадические случаи. Причем последние составляют до 70%.
Впервые туберозный склероз был описан Реклингхаузеном в 1862 г. Француз Бурневилль в 1880 г. подробно исследовал морфологические изменения, происходящие в головном мозге при этом заболевании, и впервые употребил термин «туберозный склероз». В 1890 г. дерматолог Прингл сделал описание ангиофибром лица у пациентов с ТС. Поэтому в литературе по неврологии можно встретить синонимичное название ТС — болезнь Бурневилля-Прингла.
Причины туберозного склероза
Заболевание имеет генетическую природу. Большинство случаев обусловлено возникновением новых мутаций и лишь 30% аутосомно-доминантным наследованием генных аберраций, имеющихся у родителей. Выделяют туберозный склероз тип 1, развитие которого обусловлено мутациями в гене 34 локуса 9-й хромосомы, ответственном за кодирование гамартина, и туберозный склероз тип 2, связанный с нарушениями в 13-ом участке 16-й хромосомы, отвечающем за кодирование туберина.
Биохимические аспекты патогенеза до конца не изучены. Известно только, что в норме гамартин и туберин являются факторами подавления опухолевого роста. Морфологическим субстратом выступают разросшиеся глиальные элементы церебральной ткани, гистологически представленные гигантскими клетками с атипично увеличенными ядрами и большим числом отростков. Глиальные разрастания формируют субэпендимальные узлы, корковые туберы и специфические островки в белом веществе. Все эти образования имеют тенденцию к обызвествлению. Субэпендимальные узлы зачастую дают начало образованию гигантоклеточной астроцитомы. В 10% случаев отмечается поражение тканей мозжечка. Глиальные разрастания наблюдаются также на диске зрительного нерва и в периферических отделах сетчатки.
Симптомы туберозного склероза
Клиника, которую имеет туберозный склероз, очень вариабельна. Она включает поражение центральной нервной системы (ЦНС), дерматологические и офтальмологические проявления, новообразования внутренних органов. Дебют приходится на различные возрастные периоды, но чаще туберозный склероз манифестирует в течение первых 5-ти лет жизни. Возможны различные по тяжести варианты течения. В легких случаях пациенты имеют ряд факультативных неспецифических симптомов и зачастую не проходят диагностику на наличие ТС. Туберозный склероз в стертой форме протекает без эпиприступов, олигофрении и расстройств поведения.
Поражение ЦНС
Изменения в ЦНС выступают доминирующими проявлениями ТС. Среди них наиболее часто (в 80-90% случаев) встречается судорожный синдром, с которого обычно манифестирует заболевание. Для эписиндрома, дебютирующего на первом году жизни, характерны инфантильные спазмы (синдром Веста), затем трансформирующиеся в синдром Леннокса-Гасто. Возможны атипичные абсансы, сомато- и сенсомоторные пароксизмы, вторично-генерализованные приступы. Возникновение в возрасте до года, высокая частота и гетерогенность приступов сопровождаются их резистентностью к антиконвульсантной (противоэпилептической) терапии. Эпилептические пароксизмы являются причиной задержки психического развития и нарушений поведения (агрессивности, аутизма, СДВГ) у детей.
В половине случаев туберозный склероз сопровождается выраженной в различной степени олигофренией. Наряду с эпилепсией, причиной ее развития считается наличие корковых туберов. Уже в младшем возрасте у детей отмечается анормальное поведение: общее беспокойство, капризность и недовольство наряду с медлительностью, затруднениями переключаемости внимания. Степень этих нарушений тем выше, чем раньше возник туберозный склероз. У большинства пациентов также наблюдаются нарушения сна. Они характеризуются ночными пробуждениями, инсомнией, сомнамбулизмом, ранним утренним переходом от сна к бодрствованию.
Дерматологические симптомы
Изменения со стороны кожи сопровождают туберозный склероз практически в 100% случаев. Они характеризуются большим полиморфизмом элементов и их сочетаний. Чаще всего (в 90% случаев) наблюдаются пятна гипопигментации, которые возникают обычно в первые 3 года жизни и в дальнейшем увеличивают свое количество. Они асимметрично разбросаны по ягодицам, туловищу и на передне-латеральных поверхностях конечностей. Возможна депигментация ресниц, бровей и волос. В 14% случаев выявляются участки гиперпигментации в виде пятен, более характерных для нейрофиброматоза. Как правило, их насчитывается не более 5 штук.
Ангиофибромы лица по различным данным отмечаются у 50-90% пациентов и образуются в основном после 4-летнего возраста. Это множественные или одиночные плотные узелки в виде зерен проса, красноватого или желтоватого цвета. «Шагреневая кожа» имеет место в 21-68 % случаев. Обычно возникает в период от 10 до 20 лет. Представляет собой асимметричные участки жесткой огрубевшей кожи, локализующиеся на спине и пояснице, имеющие размер от 2-3 мм до 10 см. При дерматоскопии видно, что шагреневые участки состоят из множества фиброзных гамартом.
В 25% случаев туберозный склероз сопровождается образованием фиброзных бляшек, в 30% случаев — мягких дерматофибром. До 50% больных после пубертата имеют склонные к прогредиентному росту околоногтевые фибромы. Последние более часто располагаются на ступнях. Имеют вид тусклых красных узелков или папул, окружающих ногтевую пластинку.
Офтальмологические симптомы
Отмечаются редко, хотя почти у половины больных ТС выявляется наличие гамартом зрительного нерва и/или гамартом сетчатки. Гамартомы могут иметь плоскую гладкую, незначительно возвышающуюся поверхность или представляют собой узловатое образование, иногда встречаются гамартомы смешанного типа — узловатые в центре. Основным проявлением гамартом является прогрессирующее падение зрения, но зачастую наблюдается их субклиническое течение. Возможны и другие офтальмологические расстройства: депигментация радужки, отек диска зрительного нерва, колобома, косоглазие, ангиофибромы век, катаракта.
Поражение внутренних органов
Новообразования соматических органов, сопровождающие туберозный склероз, отличаются множественностью и частым двусторонним поражением парных органов, длительно протекают субклинически. Период их манифестации колеблется от 5 до 40 лет. К наиболее патогномоничным для ТС новообразованиям относятся: рабдомиома сердца, кисты легких, поликистоз почек, гамартомы печени, ректальные полипы. В 4,5% случаев при ТС наблюдаются злокачественные опухоли, чаще почечно-клеточный рак.
Со стороны сердечно-сосудистой системы выявляются опухоли сердца. В 30-60% случаев это рабдомиомы. При их внутриутробном развитии может наблюдаться антенатальная гибель плода. У половины новорожденных с ТС рабдомиомы выявляются случайно при выполнении ЭхоКГ. У маленьких детей они проявляются аритмией, синдромом WPW, тахикардией, фибрилляцией желудочков. Интрамуральное положение рабдомиомы влечет за собой расстройство сократимости; обтурация опухолевой массой сердечных камер приводит к сердечной недостаточности. У старших детей рабдомиомы преимущественно бессимптомны; возможна блокада ножки пучка Гиса, псевдоишемические отклонения на ЭКГ. Зачастую наблюдается регресс и даже полное исчезновение рабдомиомы к 6-летнему возрасту.
Поражение легких отмечается у пациентов, имеющих туберозный склероз, после 30 лет. На рентгенограмме определяется характерная для множественных легочных кист картина «сотового легкого». Поражение ЖКТ включает опухоли полости рта, дефекты зубной эмали, множественные или одиночные гамартомы в печени, не склонные к малигнизации полипы прямой кишки. Поражение почек сопровождают туберозный склероз в 50-85%. Могут отмечаться ангиомиолипомы, кисты, гломерулосклероз, нефрокальциноз, интерстициальный нефрит, гломерулонефрит. Патология почек выступает второй после поражения ЦНС причиной летального исхода при ТС.
Диагностика
Диагностировать туберозный склероз возможно лишь совместными усилиями нескольких специалистов (невролога, офтальмолога, дерматолога, кардиолога, нефролога) с проведением широкого аппаратного обследования пациента. Церебральная эпилептическая активность регистрируется при помощи ЭЭГ и ЭЭГ с пробами. У детей до года возможно проведение нейросонографии. Наибольшую значимость в диагностике поражений ЦНС имеют КТ и МРТ. КТ головного мозга более информативно в отношении кальцифицированных туберов и субэпендимальных узлов, а МРТ головного мозга — в выявлении некальцифицированных туберов. С целью своевременной диагностики астроцитомы детям, имеющим туберозный склероз, рекомендовано прохождение МРТ или КТ-исследования не реже чем раз в 2 года.
Проводится комплексное обследование соматических органов: ЭКГ, УЗИ и МРТ сердца, УЗИ брюшной полости, УЗИ и КТ почек, урография, обзорная рентгенография грудной клетки, ректороманоскопия, колоноскопия. Диагностика офтальмологических поражений осуществляется путем прямой и непрямой офтальмоскопии, сканирующей томографии сетчатки.
В связи с большой полиморфностью сопровождающих туберозный склероз проявлений, для установления диагноза используют диагностические критерии, разработанные в 1998 г. в Швеции. Они включают первичные, вторичные и третичные признаки. Туберозный склероз достоверен, когда имеет место 1 первичный признак в сочетании с 2 вторичными или третичными. Туберозный склероз вероятен при наличии 1 вторичного и 1 третичного или 3 третичных признаков. Окончательную точку в вопросе диагностики туберозного склероза помогает поставить генетический анализ.
Лечение туберозного склероза
Основополагающим направлением в лечении ТС является антиконвульсантная терапия, поскольку степень олигофрении и ЗПР напрямую коррелирует с частотой эпиприступов, а эпилептический статус может стать причиной смертельного исхода. Выбор препарата зависит от вида пароксизмов, при недостаточной эффективности монотерапии, назначается комбинированное лечение. При синдроме Веста применяют вигабатрин и тетракозактид. Препаратами второй очереди выступают вальпроаты. Если туберозный склероз протекает с парциальными эпиприступами, то базовой терапией считается сочетание вальпроатов с карбамазепином. При отсутствии эффекта в эту схему лечения включают ламотриджин. При генерализованных эпиприступах и парциальных пароксизмах в качестве монопрепарата и в комбинации с другими противоэпилептическими средствами могут применяться современные антиконвульсанты топирамат и леветирацетам.
Терапия олигофрении проводится преимущественно путем нейропсихологической коррекции и комплексного психологического сопровождения ребенка. Назначение ноотропов и прочих стимулирующих нейропрепаратов противопоказано из-за наличия эписиндрома. При выявлении астроцитомы проводится динамическое наблюдение. Хирургическое удаление внутримозговой опухоли показано только при резком увеличении ее размеров с подъемом внутричерепного давления. Операцию проводят нейрохирурги.
В отношении новообразований соматических органов применяется преимущественно выжидательная тактика. Хирургическое лечение проводится по показаниям, в основном в случаях, когда опухоль вызывает существенную дисфункцию органа или имеется угроза ее злокачественного течения.
Факоматозы
Факоматозы — это группа прогрессирующих генетически детерминированных патологий, проявляющихся полиморфными симптомами с преимущественным поражением нервной системы, кожи, органа зрения и сопровождающихся появлением различных новообразований висцеральных органов. Диагностировать факоматоз возможно усилиями многих специалистов и только путем комплексного обследования пациента (МРТ, КТ, УЗИ, ЭКГ, ЭЭГ, офтальмоскопия и пр.). Лечение факоматоза симптоматическое: антиконвульсантное, нейрометаболическое, дегидратационное, психотерапевтическое, хирургическое. Прогноз зависит от формы и тяжести факоматоза.
Отдельные клинические варианты факоматозов были описаны различными авторами в конце XIX столетия. Впервые объединить в одну группу заболевания с сочетанным поражением нервной системы, глаз и висцеральных органов, сопровождающиеся кожными проявлениями в виде гипо- и гиперпигментаций, нейрофибром и ангиом, предложил в 1923 году нидерландский офтальмолог Ван дер Хуве. Он же ввел определение «факоматоз».
В настоящее время термин «факоматоз» не является нозологической единицей и не может быть диагнозом, он лишь обозначает принадлежность заболевания к определенной группе патологий. К наиболее распространенным нозологиям этой группы относится нейрофиброматоз Реклингхаузена и туберозный склероз. Всего группа включает около 30 нейрокожных синдромов, в том числе синдром Луи-Бар, болезнь Гиппеля-Линдау, синдром Бонне-Дешома-Блана, синдром Стерджа-Вебера, ангиоматоз Ван-Богарта-Диври, гипомеланоз Ито, синдром недержания пигмента.
Причины факоматоза
Факоматоз — это генетическое заболевание, обусловленное нарушениями в процессах дифференцировки и развития клеток в раннем эмбриональном периоде. С развитием генетики и методов ДНК-анализа для некоторых заболеваний были установлены гены, аберрации в которых детерминируют данный факоматоз. Зачастую мутации приводят к снижению синтеза факторов, блокирующих онкогенез, что считают вероятной причиной множественного опухолевого роста, в большинстве случаев характеризующего факоматоз. Исследования показали, что факоматозы имеют в основном аутосомно-доминантное наследование с неполной пенетрантностью, благодаря которой болезнь проявляется не в каждом поколении.
Патогенез
Нарушения касаются преимущественно эктодермального зародышевого листка, который дает начало всей нервной ткани, наружным слоям кожных покровов, придаткам кожи (ногтям, волосам), сетчатке, эпителию слизистой рта и полости носа. Клетки, которые остались в фазе перманентной эмбрионизации, т. е. не продолжили свое развитие, образуют врожденные опухолевые образования — гамартомы. Эти эмбриональные опухоли различной локализации часто сопровождают любой факоматоз.
В связи с тем, что факоматоз детерминируется преимущественно сбоем в развитии эктодермальных структур, в литературе по генной патологии, педиатрии и неврологии он часто носит название нейроэктодермальная дисплазия. Однако нередким при факоматозе является сочетание эктодермальной дисплазии с нарушениями дифференцировки мезо- и энтодермального зародышевых листков. Проявлениями мезодермальной дисплазии служат аневризмы, ангиомы, рабдомиомы, лейомиомы, костнотканные пороки (например, дисплазия тазобедренного сустава). Наиболее распространенным клиническим симптомом энтодермальной дисплазии выступает полипоз различных отделов ЖКТ (полипы желудка, полипы кишечника, полипы прямой кишки).
Симптомы факоматоза
Типичной чертой факоматоза выступает сочетанное полиморфное поражение кожи, нервной системы и соматических органов. Причем одни клинические синдромы, чаще всего неврологические и дерматологические, являются врожденными или манифестируют в раннем детском возрасте, а другие — намного позже. В отдельных случаях факоматоз сочетается с врожденным иммунодефицитом, преждевременным старением и/или риском развития злокачественных образований.
Неврологические проявления
Морфология поражений нервной системы сводится к образованию в веществе и оболочках мозга туберов, кист, субэпендимальных узлов, нейрофибром, кальцификатов, участков глиоза, атрофии или демиелинизации; наличию врожденных аномалий питающих мозг сосудов (аневризм, АВМ, ангиом). Клинически наиболее часто наблюдается судорожный синдром, который может иметь различное течение и большую вариабельность пароксизмов. В раннем детстве зачастую отмечается синдром Веста, у детей постарше — синдром Леннокса-Гасто, генерализованные и парциальные сенсомоторные эпиприступы, абсансы.
Вследствие эпилепсии и поражения церебральных структур факоматоз зачастую сопровождается задержкой психического развития, нарушениями речи, олигофренией, анормальным поведением. Умственное недоразвитие варьирует от дебильности до идиотии, степень его выраженности прямо коррелирует с тяжестью и частотой эпиприступов. Часто наблюдаются нарушения со стороны черепно-мозговых нервов (глазодвигательные расстройства, нарушения слуха, лицевой парез и пр.), пирамидная недостаточность (чаще по гемитипу), экстрапирамидная симптоматика (атетоз, брадикинезия, гиперкинезы, тонические мышечные симптомы), мозжечковая атаксия, расстройства сна (сомнамбулизм, инсомния). Нарушения поведения носят вариативный характер: от СДВГ до аутизма.
Кожные проявления
Дерматологические изменения, сопровождающие факоматоз, обычно выявляются в первые несколько месяцев жизни. Они бывают единичные или диффузные, вариабельные по цвету и величине, как правило, асимметричные. Наиболее часто встречаются пигментные пятна, участки гипопигментации, дерматофибромы, нейрофибромы, папилломы, шагреневые бляшки, ангиомы.
Эндокринные расстройства
В ряде случаев факоматоз протекает с эндокринно-обменными расстройствами (несахарным диабетом, гипотиреозом, ожирением, задержкой или преждевременным половым созреванием) и нарушениями вегетативной сферы и трофики (ломкостью ногтей, сухостью кожи, выпадением волос).
Офтальмологические проявления
В некоторых вариантах факоматоз включает поражение органа зрения. Обычно офтальмологические проявления имеют врожденный характер или ранний дебют. Осмотр глазного дна в таких случаях может выявить ангиоматоз или гамартомы сетчатки, телеангиэктазии конъюнктивы. Возможно их инаппарантное течение или проявление в виде снижения остроты зрения.
Другие проявления
Сопутствующие факоматозу поражения соматических органов обусловлены преимущественно развивающимися в них новообразованиями. Последние обычно имеют доброкачественный характер, но склонны к рецидивированию и прогредиентному росту. Зачастую такими образованиями являются гамартомы. Кроме того, у имеющих факоматоз детей отмечается склонность к инфекционным заболеваниям, которые, в свою очередь, усугубляют течение основной патологии.
Широкая вариативность симптомов и их манифестация в различные возрастные периоды обуславливают те трудности, которые приходится преодолевать врачам, чтобы выявить и верифицировать факоматоз. Диагностический поиск осуществляется усилиями многих специалистов: невролога, педиатра, офтальмолога, дерматолога, кардиолога, нефролога, гастроэнтеролога, генетика, эндокринолога и др. При подозрении на факоматоз проводится биохимический анализ крови и мочи, генеалогическое исследование и ДНК-анализ, широкое инструментальное и нейропсихологическое обследование.
- Неврологическая диагностика.Электроэнцефалография позволяет установить характер эпиактивности головного мозга. Эхо-ЭГ выявляет признаки гидроцефалии. При помощи МРТ и КТ головного мозга визуализируются морфологические изменения церебральных тканей, при помощи ангиографии головного мозга или МРА — пороки церебральных сосудов.
- Офтальмологическая диагностика.Офтальмоскопия проводится в обязательном порядке, позволяет диагностировать поражение органа зрения даже в случае его субклинического течения.
- Исследование внутренних органов. Кардиологические исследования включают ЭКГ и УЗИ сердца, гастроэнтерологические — УЗИ брюшной полости, при необходимости рентгенографию желудка, рентгенконтрастное обследование тонкого и толстого кишечника. Исследование почек проводится при помощи УЗИ, урографии и КТ.
Лечение факоматоза
Медикаментозная терапия
На сегодняшний день ни один факоматоз не имеет специфического лечения. Проводится симптоматическая терапия. По показаниям применяется антиконвульсантное (вальпроаты, леветирацетам, карбамазепин, топирамат), дегидратирующее (ацетазоламид), нейрометаболическое (витамины группы В, глицин) лечение. Зачастую эпиприступы оказываются резистентными к проводимой противосудорожной терапии, в связи с чем приходится менять препарат или переходить на комбинированные схемы, включающие 2 антиконвульсанта. При наличии эписиндрома противопоказаны нейрометаболиты стимулирующего действия (к-та гамма-аминомасляная, пирацетам, пиритинол).
Хирургическое лечение
По показаниям проводится хирургическое лечение, целью которого является удаление возникшего новообразования. Вмешательства проводятся при подозрении на злокачественность опухоли, при нарастании обусловленных ею клинических проявлений, быстром росте образования, развитии компрессионного синдрома. Если речь идет о внутримозговых опухолях, то операцию проводят нейрохирурги. При опухолях соматических органов оперируют соответствующие специалисты.
Психологическая коррекция
Наряду с фармакотерапией в лечении факоматоза большую роль играет психокоррекция. Она направлена на развитие умственных и психических способностей ребенка, индивидуальную коррекцию имеющихся отклонений, обучение ребенка в доступном для него формате и его социальную адаптацию. В зависимости от вида и степени психических нарушений рекомендованы занятия с психологом, детская психотерапия, игровая терапия, АВА терапия, нейропсихологическая коррекция. Осуществляется психологическое консультирование родителей. Возможно комплексное психологическое сопровождение ребенка.
Прогноз и профилактика
В основном факоматозы относятся к прогностически неблагоприятным заболеваниям. Исход зависит от типа факоматоза, возраста, в котором он дебютировал, и тяжести патологии. Усугубить течение факоматоза может инфекционное заболевание или травма.
Прогностически неблагоприятными факторами выступают: ранняя манифестация, тяжелый эписиндром, глубокая олигофрения, развитие злокачественных новообразований. Гибель пациентов происходит при возникновении отека головного мозга, эпилептического статуса, раковой кахексии, сепсиса вследствие интеркуррентной инфекции.
Основным средством предупредить факоматоз является исключение возможности рождения больного ребенка. С этой целью проводится генетическое консультирование пар, планирующих беременность. Вероятность рождения ребенка с факоматозом является основанием для ограничения деторождения.
Генодерматозы
Генодерматозы — это общее название неоднородной группы наследственных заболеваний с преимущественным поражением кожи. Они характеризуются значительным полиморфизмом клинических проявлений, подразделяются на 6 групп соответственно фенотипическим признакам и основному механизму развития. К специфическим симптомам относят очаги гипер- или гипопигментации, участки повышенного ороговения и чешуйчатости кожи, накожные опухолевые образования. План диагностики состоит из клинических, биохимических, гистологических и молекулярно-генетических методов. Лечение поддерживающее, определяется типом генодерматоза, тяжестью симптоматики.
Насчитывается около 200 генетически обусловленных кожных болезней, которые составляют 35% от наследственных нозологий, до 10% от всех дерматологических заболеваний. Поражение кожного покрова может быть единственным проявлением патологии, но чаще оно возникает в комплексе с мультиорганными нарушениями. Генодерматозы представляют серьезную проблему в современной дерматологии, что вызвано сложностью их лечения, отсутствием этиопатогенетической терапии, повышенным риском появления опухолей при ряде кожных заболеваний.
Причины генодерматозов
Истинные генодерматозы имеют моногенную природу — развиваются при мутации определенного гена в соматических или половых хромосомах. По характеру наследования на первом месте находится аутосомно-доминантный вариант, когда ребенку для развития болезни достаточно получить генную аномалию от одного родителя, а риск возникновения синдрома составляет 50% для младенцев обоего пола. Реже встречаются аутосомно-рецессивный, Х-сцепленный типы наследования.
При мультифакториальных генодерматозах для манифестации процесса необходимы экзогенные провоцирующие воздействия. К распространенным триггерам относят эндокринные нарушения (патологии гипоталамуса, эпифиза, щитовидной железы), хронические очаги инфекции (тонзиллит, кариес, риносинуситы), аллергические расстройства. Важными провоцирующими факторами являются повышенные психоэмоциональные нагрузки, неблагоприятная экологическая ситуация.
Учитывая большую вариабельность заболеваний, входящих в группу генодерматозов, выделить типичные патогенетические особенности не представляется возможным. В основе большинства пигментных нарушений лежит повышенная чувствительность клеток кожи к УФ-лучам, на фоне чего формируются очаги гиперпигментации. Генодерматозы, проявляющиеся гипопигментацией, возникают при снижении или отсутствии выработки меланина вследствие мутаций соответствующих ферментов.
Ихтиозы характеризуются нарушениями процессов кожного ороговения, замедлением отшелушивания отмерших кератиноцитов, что сопровождается утолщением и изменением внешнего вида кожных покровов. При опухолевых кожных синдромах патогенез связан с мутациями генов, отвечающих за подавление роста новообразований, в результате чего больные сталкиваются с множественными доброкачественными неоплазиями.
Классификация
В отечественной медицинской литературе распространено разделение заболеваний на 10 групп по локализации и характеру поражения. В международной дерматологической практике чаще используется классификация с учетом механизма развития и фенотипических проявлений генодерматозов, согласно которой выделяют следующие формы:
- Генодерматозы с нарушением репарации ДНК: пигментная ксеродерма, синдром Вернера, синдром Ротмунда-Томсона.
- Генодерматозы с нарушенной кератинизацией: ихтиоз, врожденный дискератоз, синдром эпителиального невуса.
- Генодерматозы с лентигинозной гиперпигментацией: синдром Пейтца-Егерса-Турена (гамартомный полипоз ЖКТ).
- Генодерматозы с кожной гипопигментацией: альбинизм, синдром Германски-Пудлака.
- Буллезные генодерматозы: буллезный эпидермолиз.
- Наследственные опухолевые синдромы: нейрофиброматоз, туберозный склероз, синдромы Кауена, Гарднера, Мюира-Торре.
В отдельную категорию выделяются мультифакториальные заболевания, которые характеризуются наследственной предрасположенностью, но для их манифестации требуется негативное влияние внешних факторов. К этой группе относят псориаз, атопический дерматит, витилиго. Большую гетерогенную группу представляют врожденные нарушения обмена веществ, которые сопровождаются кожным поражением, — болезнь Нимана-Пика, фенилкетонурия, гомоцистинурия.
Симптомы генодерматозов
Большинство заболеваний проявляются в первые месяцы после рождения ребенка. Настораживающими признаками являются патологии вынашивания беременности, преждевременные роды, симптомы задержки внутриутробного развития плода. Отдельные нозологии, например, болезнь Вернера, манифестируют у взрослых, пик их диагностики приходится на возраст 20-30 лет.
Генодерматозы с пигментными нарушениями
Для генодерматозов, протекающих с гиперпигментацией, характерны участки фотодерматита, веснушки и крупные коричневые пятна на открытых зонах тела ребенка, сухость и гиперкератоз. При прогрессировании процесса появляются участки атрофии, депигментации, деформации ушных раковин, кончика носа. У страдающих альбинизмом, напротив, отмечается неестественная бледность кожи, красноватый оттенок зрачков, отсутствие пигмента в волосах, ресницах, бровях.
Генодерматозы с нарушениями кератинизации
Генодерматозы из группы ихтиоза имеют патогномоничные проявления: обилие плотных чешуек на кожных покровах, которые придают вид змеиной кожи. Чешуйки имеют темно-коричневый цвет, плотно связаны с эпидермисом, практически не удаляются при гигиенических процедурах и трении мочалкой. Врожденный дискератоз отличается дистрофическими изменениями ногтей, лейкоплакией слизистых оболочек.
Опухолевые генодерматозы
При факоматозах (опухолевых синдромах) характерным является сочетание кожных признаков с поражением нервной системы. На теле появляются множественные болезненные новообразования темно-розового или коричневого оттенка, достигающие диаметра несколько сантиметров. По ходу нервных стволов также прощупываются многочисленные плоские опухоли. Патогномонична кожная пигментация в виде молочно-кофейных пятен.
Осложнения
Генодерматозы, как правило, сопровождаются поражением глаз по типу блефарита, кератита, эктропиона. Реже наблюдаются изъязвление роговицы, папилломы, базальноклеточный рак века. Наиболее опасным осложнением большинства опухолевых и пигментных кожных изменений является высокий риск малигнизации — образования злокачественных неоплазий. Они являются основной причиной смерти больных с генодерматозами.
При части наследственных дерматозов, например, гамартомном полипозе ЖКТ в процесс вовлекаются многие внутренние органы. Такие пациенты страдают от мультисистемных нарушений, определяющих тяжесть состояния. Обычно нозологии сочетаются с нервно-психическими расстройствами: у детей возникает синдром дефицита внимания и гиперактивности (СДВГ), расстройства аутистического спектра, проблемы с социализацией из-за нетипичной внешности.
Основу постановки диагноза составляет клинический осмотр пациента у детского или взрослого дерматолога. При выявлении типичных признаков (участки гипопигментации, плотные чешуйки, буллезные высыпания) удается установить предварительный диагноз, чтобы целенаправленно проводить лабораторно-инструментальную диагностику проблемы. Затем назначается комплекс диагностических мероприятий:
- Гистология кожи. Основной метод исследования, который применяется для изучения микроструктуры кожного покрова, обнаружения избыточных пигментных клеток, гиперкератоза, истончения росткового слоя. При большинстве болезней определяется отечность, воспалительная инфильтрация дермы.
- Дополнительные инструментальные методы. Зачастую генодерматозы комбинируются с соматической патологией. Поэтому больным показаны визуализационные исследования. При неопластических синдромах обязательно проводится КТ или МРТ головного мозга. Для оценки структурно-функциональных особенностей ЖКТ рекомендуются УЗИ органов брюшной полости, рентгенография.
- Биохимические анализы. При кератозах исследуется активность STS в лейкоцитах, выполняется электрофорез сывороточных протеинов для дифференцировки разных форм ихтиозов. Ценную информацию в уточнении причины генодерматозов играют иммунофлюоресцентные, иммуногистохимические методики.
- Генетическое тестирование. Обследование генома пациента — самый точный диагностический метод. Применяются методики секвенирования экзона или генома, флюоресцентной гибридизации (FISH). Поскольку исследования дорогостоящие и трудоемкие, они назначаются по строгим показаниям при невозможности установить диагноз другими способами.
Ряд моногенных кожных патологий подлежит диагностике еще во внутриутробном периоде, если родители больны или являются носителями мутантных генов. Для пренатального обнаружения генодерматозов используются ультраструктурный анализ биоптатов кожи плода, определение активности галактозидазы в амниотической жидкости, исследование уровня стероидной сульфатазы. При необходимости производится генотипирование биоматериала после амниоцентеза, биопсии хориона.
Лечение генодерматозов
Поскольку патологии вызваны мутациями в генетическом аппарате клеток, этиопатогенетического лечения не существует. Заболевания требуют пожизненной поддерживающей терапии, чтобы контролировать интенсивность кожных проявлений, нормализовать самочувствие больных, по возможности устранить неэстетичные очаги на коже. Программа лечения включает следующие направления:
- Подбор плавильной косметики. Для ухода за поврежденной кожей используются средства с кератолитиками, смягчающими и питательными компонентами, органическими кислотами. При некоторых болезнях рекомендованы топические стероиды, местные ретиноиды.
- Защита от солнца. Ультрафиолет является основным триггером развития злокачественных опухолей кожи при генодерматозах, поэтому в теплое время года обязательно наносить кремы с SPF 30-50 на всех открытых участках тела. При альбинизме нужно носить контактные линзы или очки, блокирующие УФ-лучи.
- Малоинвазивные методики. При доброкачественных опухолевых образованиях возможно их удаление с помощью электрокоагуляции, криодеструкции, лазерного излучения. Для улучшения состояния кожных покровов может назначаться дермабразия и другие косметологические процедуры.
Хотя генодерматозы невозможно полностью вылечить, многие из них удается успешно контролировать поддерживающей терапией, чтобы минимизировать клинические симптомы, улучшить качество жизни больных. Менее благоприятен прогноз для пациентов с неопластическими синдромами, которые часто заканчиваются смертью от злокачественных новообразований, а также отягощении генодерматоза тяжелыми метаболическими или соматическими системными патологиями.
Учитывая наследственный характер болезней, основу профилактики составляет медико-генетическое консультирование пар из групп риска в периоде планирования зачатия. При наличии сведений о тяжелых семейных генодерматозах целесообразно проводить пренатальный скрининг либо предимплантационную генетическую диагностику (при прохождении протокола ЭКО). Для предупреждения осложнений требуется пожизненный особый уход за кожей, диспансерное наблюдение у дерматолога.
1. Факоматозы: диагностика, клиника и особенности течения различных форм заболевания/ Л. А. Юсупова, Е. И. Юнусова, З. Ш. Гараева, Г. И. Мавлютова// Лечащий врач. — 2018. — №5.
2. Федеральные клинические рекомендации. Дерматовенерология 2015: Болезни кожи. Инфекции, передаваемые половым путем. — 2016.
3. Подходы к формированию генетического прогноза развития мультифакториальных заболеваний/ А.М. Федота// Вестник биологии и медицины. — 2014. — №4.
Группа нейрокожных заболеваний факоматозы: симптомы и лечение
На сегодняшний день известно большое количество наследственных заболеваний, поражающих спинной, головной мозг и периферические нервные волокна вследствие генетических мутаций, передаваемых в поколениях по вертикали.
Нервная система человека отличается чрезвычайной сложностью и уязвимостью. Любые неблагоприятные влияния на плод, особенно на гены, ответственные за разные структуры нервной системы, могут спровоцировать появление тяжелой патологий.
Например, нарушение нормального развития листков эктодермы и мезодермы у зародыша приводит к поражению нервной ткани, кожи, глаз, сосудов и внутренних органов. Такая мультисистемность характерна для факоматозов. Что это за патология — попробуем разобраться.
Особенности группы заболеваний
Впервые факоматозы были описаны как большая группа заболеваний наследственного характера в 20-е годы прошлого столетия. В эту группу объединили патологические состояния, связанные с изменениями кожных покровов в комбинации с поражением органов зрения, разных отделов нервной системы и внутренних органов (всего более 50 нозологических форм).
Клинически факоматозы обнаруживаются уже в раннем возрасте или сразу после рождения, обычно это дерматологические проявления. В то же время неврологические симптомы могут появиться значительно позднее — в подростковом и зрелом возрасте. В отличие от врожденных пороков развития данная патология имеет неуклонно прогрессирующее течение.

В основе всех видов заболевания лежит мутация генов, отвечающих за выработку супрессора (фактора подавления) роста опухолей.
Наследование патологии происходит по вертикали от родителей к детям по аутосомно-доминантному типу (половина детей получат аномальный ген от матери или отца).
Эти болезни отличаются значительной гетерогенностью — наряду с тяжелыми прогностически неблагоприятными формами наблюдаются стертые моносимптомные варианты.
Общим для всех видов факоматозов является наличие множественных опухолевых разрастаний — это гемангиомы, фибромы, папилломы, телеангиоэктазии и пигментные пятна на коже. В то же время каждая нозологическая форма имеет специфические особенности.
Классификация заболеваний
Термин «факоматоз» не может быть использован в качестве диагноза, он обозначает отношение патологии к определенной группе нейрокожных заболеваний, существует классификация, исходя из которой самыми распространенными формами среди них являются:
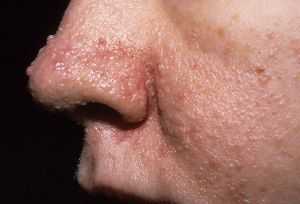
- болезнь Реклингхаузена (нейрофиброматоз 1 типа) — неизлечимая прогрессирующая патология с большим количеством узелковых разрастаний (нейрофибром), пигментными пятнами на коже кофейного цвета, опухолями нервной системы и аномалиями опорно-двигательного аппарата;
- нейрофиброматоз 2 типа — характеризуется множественными более активными опухолями, в частности двусторонней симметричной невриномой слухового нерва;
Клинические проявления
Для всех факоматозов типичным является выраженный полиморфизм кожных проявлений, поражение разных структур нервной системы и висцеральных органов. Характерно то, что в раннем возрасте выявляются в основном дерматологические симптомы, а неврологические и другие обнаруживаются позднее.
В некоторых случаях факоматоз сочетается с врожденным дефицитом иммунной системы (Луи-Бара болезнь), преждевременным развитием старения и (или) риском малигнизации опухолей.
Поражение центральной нервной системы при этой патологии морфологически выглядит, как разрастание в оболочках и ткани мозга узелков, кистозных полостей, нейрофиброматозных образований, участков демиелинизации, атрофии и кальцификации, врожденных сосудистых аномалий по типу аневризм, гемангиом.
В клинике присутствует картина судорожного синдрома с различным характером течения. У деток часто случаются эпилептические припадки, вследствие чего факоматоз нередко сопровождается задержкой нервно-психического развития, олигофренией, асоциальным поведением. Степень выраженности умственной отсталости прямо пропорциональна тяжести и частоте эпилептических приступов.
Для группы заболеваний характерны поражения разных пар черепных нервов с развитием глазодвигательных, слуховых нарушений, парезов и других симптомов.
Может присутствовать пирамидная недостаточность (чаще одностороннего характера), экстрапирамидные расстройства (бради— и олигокинезия, гиперкинезия, тонические мышечные спазмы), мозжечковые дисфункции (нарушения координации), расстройства функции сна.
Дерматологические проявления обычно выявляются в первые недели или месяцы жизни. Элементы сыпи бывают единичными или распространенными, различного цвета и размера, чаще асимметричные.
Типичными считаются пигментные пятна разного оттенка коричневого цвета, участки пониженной пигментации, узелковые элементы, нейрофибромы, папилломы, шагреневая кожа с бляшками, гемангиомы.
В некоторых случаях факоматоз протекает с эндокринными расстройствами по типу ожирения, несахарного диабета, нарушения нормального полового развития. Есть симптомы вегетативно-трофического характера (потеря волос, ломкость ногтей, сухость и шелушение кожи).
Достаточно часто бывает поражение глаз, которое проявляется достаточно рано после рождения ребенка. При осмотре выявляется гемангиоматоз сетчатки, стойкое патологическое расширение сосудов конъюнктивы. Возможно бессимптомное течение и проявление только в виде пониженной остроты зрения.
Поражения внутренних органов структурно и функционально обусловлены развивающимися в них опухолями, которые чаще имеют доброкачественный характер роста, но склонны к частому рецидивированию и прогрессированию.
Кроме того, сниженный иммунитет у детей с факоматозом способствует склонности к присоединению разных инфекций, которые ухудшают течение основного недуга.
Диагностика и терапия

Любое подозрение на факоматоз требует консультации специалистов из разных отраслей медицины — педиатрии, генетики, неврологии, эндокринологии, психиатрии, дерматологии, офтальмологии.
Из обследований проводится генеалогический и ДНК-анализ, биохимия крови и мочи, широкий спектр инструментальных и нейропсихологических исследований — ЭЭГ, Эхо-графия мозга и внутренних органов, МРТ, КТ, ангиография, рентгеновские исследования почек, желудка и кишечника. При необходимости назначаются другие виды обследований для уточнения диагноза.
Специфического лечения факоматозов нет. Проводится только симптоматическая терапия по показаниям для каждого больного с конкретной формой заболевания. Назначаются следующие группы препаратов:
- противосудорожные (Карбамазепин, Топалепсин, Эпитропил и другие);
- мочегонные (например, Диакарб);
- нейрометаболические (витамины В12, В6, Ноотропил, Энцефабол, Аминалон).
При неэффективности консервативной терапии проводится удаление новообразований хирургическим способом, которое особенно показано при подозрении на злокачественный характер опухоли, при резком прогрессировании клинических симптомов, развитии синдрома сдавления мозга.
Наряду с медикаментами в лечении факоматоза большую роль играют психокорректирующие мероприятия, направленные на восстановление или развитие нарушенных интеллектуальных способностей ребенка, купирование выявленных психических отклонений, обучение и социальную адаптированность. Практикуется комплексная психологическая помощь семьям.
Не нужно вешать нос
В большинстве случаев факоматозы имеют неблагоприятный прогноз, который напрямую зависит от вида патологии, возраста дебюта болезни и тяжести течения, а также наличия иммунодефицита, полученных травм.
Хуже всего прогноз при раннем начале заболевания, тяжелом эписиндроме, глубокой степени олигофрении, появлении злокачественных новообразований.
Основным средством профилактики является консультирование пар, планирующих зачатие ребенка, специалистом в области медицинской генетики. Возможность деторождения должна определяться индивидуально в каждом случае.
Трудности диагностики заболеваний из группы факоматозов связаны со значительным полиморфизмом клинических проявлений и зависимостью дебюта симптомов от возраста пациента.
Больные с данной патологией в течение всей жизни наблюдаются врачами многих специальностей. Вот почему имеет значение большая информированность по данному заболеванию. Выбор правильного способа и тактики ведения пациентов возможен только при согласованном действии разных специалистов.
Факоматозы - группа наследственных заболеваний, при которых происходит сочетанное поражение нервной системы, кожи, сетчатки и внутренних органов. Патология развивается в детском возрасте, но может быть диагностирована и при рождении.
Причины факоматоза

Заболевание передается по аутосомно-доминантному, реже - по аутосомно-рецессивному типу. Специалисты предполагают, что в основе факоматоза лежат генетически обусловленные нарушения процессов дифференцировки и развития зародышевых клеток в раннем эмбриональном периоде.
Благодаря современным методам ДНК-диагностики ученые смогли установить гены, мутации которых вызывают различные формы факоматозов. В большинстве случаев в основе появления патологий лежит снижение синтеза факторов, ответственных за блокировку онкогенеза, что обуславливает множественный опухолевый рост.
Патогенез
На ранних стадиях эмбриогенеза происходят нарушения в росте и дифференцировке клеток одного или нескольких эктодермальных зародышевых листков. Такие клетки «застывают» в своем развитии и образуют гамартомы - врожденные опухолевые образования, которые склонны к пролиферации и неопластической трансформации.
Наиболее частыми проявлениями эктодермальной дисплазии являются невусы, гипо- и гиперпигментированные пятна на коже, факомы на сетчатке, субэпендимальные и кортикальные узелки в ЦНС.
Эктодермальная дисплазия нередко сочетается с другими видами дисплазии - мезодермальной (аневризмы, ангиомы, рабдомиомы, лейомиомы) и энтодермальной (полипозы различных отделов пищеварительного тракта).
Симптомы факоматоза
Основу клинической картины любого факоматоза составляют поражения кожи, нервной системы, органов зрения и внутренних органов.
- Проявления со стороны нервной системы. Наиболее часто встречаются эпилептические припадки (парциальные или генерализованные). Также у пациентов отмечают задержку психического развития и нарушения речи. При вовлечении в патологический процесс черепно-мозговых нервов возможно появление глазодвигательных расстройств и проблем со слухом. Другими симптомами поражения нервной системы при факоматозах могут быть расстройства сна и двигательные нарушения (различные формы гиперкинезов, брадикинезия).
- Поражение кожных покровов. Симптоматика представлена гиперпигментированными пятнами, участками гипопигментации, шагреневыми бляшками, ангиомами, телеангиоэктазиями.
- Нарушения со стороны органов зрения. При осмотре глазного дна специалисты выявляют гамартомы сетчатки, телеангиоэктазии на конъюнктиве. Возможно снижение остроты зрения.
- Эндокринологические расстройства. Факоматозы могут сочетаться с несахарным диабетом, гипотиреозом, ожирением, нарушениями полового созревания.
- Поражение внутренних органов. Симптоматика обусловлена развивающимися в них новообразованиями, преимущественно доброкачественными, но склонными к прогрессированию и рецидивированию.
В некоторых случаях факоматозы сочетаются с врожденным иммунодефицитом, что проявляется высокой восприимчивостью к инфекционным заболеваниям, усугубляющим состояние пациентов.
Диагностика факоматоза

В диагностическом поиске участвует команда врачей: невролог, педиатр, офтальмолог, дерматолог, генетик и др. При постановке диагноза учитывают клиническую картину (сочетание эпилептических приступов, специфических изменений глазного дна и поражений кожи) и данные обследования пациента:
- Краниография. На снимках в различных структурах головного мозга видны кальцификаты - отложения солей кальция.
- Пневмоэнцефалография. Показывает увеличение желудочков мозга и признаки внутричерепной гипертензии.
- Эхоэнцефалография. Обнаруживает признаки гидроцефалии.
- МРТ и КТ головного мозга. Выявляют ангиомы, нейрофибромы, различные новообразовании и атрофические процессы.
- Ангиография головного мозга. Помогает визуализировать пороки церебральных сосудов.
- Электроэнцефалография. На энцефалограмме будут изменения биоэлектрической активности эпилептического характера.
- Офтальмоскопия. Визуализирует патологические изменения глазного дна.
При подозрении на поражение внутренних органов по показаниям проводят дополнительное обследование, включающее анализ крови, УЗИ органов брюшной полости, ЭКГ, УЗИ сердца, рентгенографию кишечника, урографию, а также КТ.
Лечение и профилактика

Специфического лечения не разработано. Медикаментозная терапия только симптоматическая. В ее схему включают противосудорожные препараты, нейрометаболиты и диуретики.
К хирургическому лечению прибегают в случаях быстрого роста опухоли, что провоцирует развитие компрессионного синдрома, а также при подозрениях на злокачественный характер новообразования.
При одиночных ангиомах кожи применяют криохирургию. Если обнаруживают ангиому мозговых оболочек, пациенту рекомендуют лучевую терапию. Ангиоретикулемы сетчатки устраняют с помощью лазера.
Поскольку заболевание проявляется в детском возрасте, ребенку обязательно требуется психокоррекция, цель которой - развитие его умственных способностей, социальная адаптация, обучение в доступной форме и коррекция имеющихся отклонений. Родителям могут быть рекомендованы различные форматы и методы психологической помощи ребенку: занятия с психологом, игровая терапия, детская психотерапия, нейропсихологическая коррекция.
Профилактика факоматозов основывается только на медико-генетическом консультировании семейной пары при планировании беременности. Также специалисты рекомендуют ограничить деторождение у пар с неблагоприятным семейным анамнезом, когда уже есть установленные случаи факоматоза у родственников.
Читайте также:
- Болезни толстой кишки
- Доброкачественная врожденная гипотония у детей. Артрогрипоз
- Сердце при сужении артериального конуса. Морфология закрытой вальвулотомии по Броку
- Диагностика поражений сердца при тиреотоксикозе. Дифференциация поражений сердца при тиреотоксикозе
- Физическая культура танцоров. Повреждения стопы у артистов балета