Гипоплазия рук, сердечная аритмия, деформация ушной раковины с глухотой. Болезнь Камурати — Энгельманна
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Микротия - это врожденная деформация ушной раковины, которая встречается в одном из 5000-7000 рождений в зависимости от той или иной статистики в разных странах и разных этнических группах. В переводе с латинского языка термин «микротия» означает «маленькое ухо». Существует 3 степени микротии, в 90% случаев вовлечена только одна сторона, правосторонняя микротия встречается в 2 раза чаще, чем левосторонняя, частота встречаемости у мальчиков 65%, у девочек - 35%.
Наследственные факторы
По данным многих исследований существуют структурные, анатомические и генетические взаимосвязи между такими патологическими состояниями, как микротия, суженные и оттопыренные уши. Показано, что эти деформации взаимосвязаны и могут быть наследственными.
Выявлены как доминантные, так и рецессивные факторы наследования при глухоте, связанной с патологией ушных раковин. Деформации ушных раковин часто встречаются в семьях с синдромом Тричер-Коллинза (мандибулофациальный синостоз).
В исследовании семей с микротией у одного или обоих родителей с исключением хромосомных аберраций пришли к выводу, что наследование должно быть мультифакторным и что риск рецидива составляет в среднем 5,7% (от 3 до 8%) [95]. Среди ближайших родственников помимо различных дефектов и деформаций ушной раковины наблюдалось нарушение развития челюстей и лицевого нерва.
Специфические факторы
Среди ученых есть предположение, что ишемия тканей (снижение кровотока) вследствие закупорки артерии может стать причиной патологического развития ушных раковин. Это говорит о том, что причиной развития деформации чаще являются проблемы, возникающие во время развития плода, а не наследственные факторы. В поддержку этой теории выступает факт частого наличия микротии только у одного из близнецов. Детские дизморфологи заявляют, что многие виды врожденных уродств чаще встречаются при многоплодной беременности, и что это может быть вследствие явления под названием синдром "фетоплацентарного обкрадывания", когда плацента нормального двойника часто больше, чем плацента аномального, и, возможно, происходит усугубление кровообращения одного из близнецов, развитие которого затем нарушается.
Хорошо известно, что появление глухоты и иногда микротии происходит в результате краснухи в течение первого триместра беременности. Кроме того, использование некоторых лекарств в этот критический период тоже может стать причиной врожденных заболеваний ( например, талидомида, роаккутан (Accutane), кломид, ретиноевая кислота).
Влияние различных внешних факторов во время первого триместра беременности на развитие микротии у ребенка не однозначно, в 1-4% случаев это могут быть простудные заболевания, корь, травмы, облучение, месячные во время первого триместра, гиперемезис (чрезмерная рвота), наличие диабета, эмоциональные перегрузки, употребление алкоголя, лекарства от тошноты.
В заключение, развитие микротии - это, как правило, случайное спорадическое событие, и важно, чтобы родители понимали, что деформация не была вызвана какими-либо действиями матери до или во время беременности, и что риск рецидива в семье составляет около пяти процентов, или, иначе говоря, один из двадцати.
Ассоциированные патологии
Эмбриологическое развитие подразумевает, что микротия обычно сопровождается патологией среднего уха. При классической микротии обычно присутствует атрезия слухового канала и нарушения слуховых косточек - Синдром Конигсмарка (микротия, атрезия наружного слухового прохода и кондуктивная тугоухость).
Деформация среднего уха может варьироваться от суженного канала и незначительных нарушений слуховых косточек до расплавленных гипоплазированных слуховых косточек и недостаточной аэрации сосцевидной кости. Следует обратить внимание, что, поскольку у пациентов с атрезией есть слуховые трубы, как и у всех, они могут спровоцировать воспаление среднего уха (средний отит) даже если они не имеют наружный слуховой проход. Поэтому, если этот диагноз невозможно подтвердить с помощью отоскопии, при наличии подозрения на средний отит в деформированном ухе, разумно назначить антибиотики.
Патология челюстно-лицевой области
Так как ушная раковина развивается из тканей жаберных дуг, не удивительно, что у значительного процента пациентов с микротией обнаруживают дефицитные компоненты лица, которые происходят из этих эмбриональных элементов. Проявление в виде уменьшенной половины лица, состояние, известное как гемифациальная микросомия - это, в основе, недоразвитие челюстей и вышележащих мягких тканей. Наиболее полное генетическое проявление этого состояния включает в себя дефекты наружного и среднего уха, гипоплазию верхней и нижней челюстей, скуловой и височной костей, макростому и боковые щели лица, парез лицевого нерва, атрофии мышц лица и околоушной железы, даже небные мышцы могут быть ослаблены на вовлеченной стороне.
Почки и мочевыводящие пути
Нарушения урогенитального тракта увеличиваются при наличии деформаций ушной раковины, особенно когда пациент страдает от других проявлений недоразвития лица. У некоторых пациентов может наблюдаться недоразвитие половых органов (гипоспадия, агенезия женских половых органов), различные нарушения почек (подковообразная почка, односторонняя почечная агенезия, тазовое расположение почки и т.п.). Однако эти нарушения не вызывают жизненно-опасных нарушений работы органов мочеполовой системы. Рутинный анализ мочи может обнаружить скрытую гематурию или протеинурию, но чаще ничего не показывает. При повторяющихся инфекциях мочевых путей у пациентов с микротией исследование функции почек следует начать с УЗИ почек, прежде чем применять более инвазивные методики для выявления патологии. Из-за повышенной заболеваемости органов мочеполовой системы у больных с микротией целесообразно проводить периодическое обследование пациентов с помощью ультразвука.
Шейный отдел (шея) позвоночника
Аномалии шейного отдела позвоночника чаще встречаются у пациентов с микротией, если присутствуют т.н. «дефекты срединной линии», например, расстройства сердца или почек или расщелины губы и неба. Так как связанные неврологические симптомы - это редкое явление, частота этих аномалий достаточно низкая у пациентов с микротией. Синдром Гольденхара (окулоаурикуловертебральная дисплазия) - состояние, при котором дефект ушной раковины сочетается с наличием дермоидной кисты глаза и обычно присутствует аномалия развития шейного отдела позвоночника. Если у пациента с микротией отмечается наличие дермоида в глазу или ограничение движения шеи, необходимо исследовать функцию почек и оценить состояние шейного отдела позвоночника.
Если есть какие-либо подозрения на наличие неврологических нарушений необходимо пройти КТ, МРТ и соответствующее неврологическое обследование.
Другие патологии: расщелина губы и неба, сердце
У небольшого процента пациентов встречаются расщелины губы и неба и пороки сердечно-сосудистой системы. Последнее включает в себя дефект межпредсердной и межжелудочковой перегородок сердца, декстрокардию, транспозицию магистральных сосудов, трехкамерное сердце и незаращенный протока. Если отмечены какие-либо признаки или симптомы этих проблем с сердцем, педиатр должен проконсультироваться с кардиологом для соответствующего ведения пациентов.
Варианты микротии
Микротия варьируется от полного отсутствия ушных тканей (anotia) до практически нормального, но маленького, уха с суженным каналом. Между этими крайностями, каждый находит бесконечное разнообразие видов, наиболее распространенным из которых является вертикально-ориентированный бобовидной формы комочек. По данным статистики, микротия почти в два раза чаще встречается у мужчин, чем у женщин, соотношение правосторонней - левосторонней и двусторонней микротии составляет примерно 6: 3: 1.
В большинстве случаев, мочка дефектного уха смещается выше уровня противоположной нормальной стороны, но при этом при развитии происходит миграция пораженного уха в более низкое положение. Примерно одна треть пациентов имеют признаки гемифациальный микросомии, но учеными при обследовании пациентов с помощью рентгеновских исследований было показано, что недоразвитие костной ткани встречается во всех случаях.
Различают 3 степени микротии (I ст. - уменьшенная ушная раковина, II ст. - недоразвитие некоторых структур ушной раковины, III ст. - ушная раковина представлена в виде бобовидного комочка) и полное отсутствие ушной раковины - анотия.
Психическое здоровье и ушная раковина
По моему опыту, ребенок узнает, что он / она отличается от других примерно в возрасте до 3-3,5 лет. В классической ситуации родители находят своего ребенка у зеркала, сравнивающего разные стороны лица. Они начинают называть дефектное ухо, как "маленькое ухо" или "закрытое ухо". Лучше всего согласиться с ребенком, что он родился с одним большим ухом и одним маленьким, а когда он подрастет, можно будет маленькое ушко сделать больше, соответствующим другому. Затем дети должны воспитываться абсолютно нормально, не делая акцента на деформацию. Дети выглядят обеспокоенными по поводу микротии до 6-7 лет только в том случае, если родители передают свои опасения ребенку.
Первое большое психологическое потрясение происходит примерно в первом классе школы, когда дети впервые попадают в большую группу своих сверстников. Именно в это время, когда уровень самосознания повышается, мы начинаем сравнивать себя друг с другом и формируем реальное представление о своем теле. Это время, когда начинаются подразнивания и издевки, и пациент с микротией узнает, что значит «быть другим».
"Второй тур" начинается с подросткового возраста, когда каждый подвергается давлению со стороны сверстников и стремится быть принятым. Это период нашей жизни, когда внешний вид становится очень важным, и каждый хочет «вписаться». Подростки с микротией очень застенчивы, так как отличаются от других, и особенно мотивированы к исправлению ушной раковины. Тем не менее, они также очень требовательны и могут иметь нереалистичные ожидания того, что может быть достигнуто хирургическим путем.
Исходя из моего опыта, нелеченный человек никогда не теряет желания стать полноценным и иметь исправленное ухо; на сегодняшний день самый взрослый пациент, которому я оперировала ушную раковину, 54 года.
В дополнение к таким функциям, как поддерживание очков, направление звуковой волны к барабанной перепонке, чтобы улучшить слух, ушные раковины позволяют нам выглядеть лучше и чувствовать себя как целостная личность. Это и является движущей причиной для хирургического создания наружного уха. Это психоэмоциональное эстетическое стремление восстановить самооценку путем восстановления симметричного, нормального образа. Совсем не являясь "косметической хирургией", исправление врожденной деформации позволяет человеку иметь нормальный образ самого себя, нормальную жизнь, и быть нормальным, продуктивным членом общества.
Если же наружное ухо не исправляется или достигается плохой результат вследствие отсутствия достаточного опыта у хирурга, состояние пациентов часто усугубляется, и страдание от низкой самооценки может длиться всю жизнь.
Из-за этих последствий, очень важно, чтобы педиатры направляли семью к опытному хирургу, даже если необходима далекая поездка для получения специализированной оценки.
Возраст для начала хирургического лечения.
Возраст, при котором следует начинать формирование ушной раковины, определяется как психологическими, так и физиологическими факторами. Восприятие изображения тела, как правило, начинает формироваться в возрасте около четырех-пяти лет, поэтому, в идеале, начать хирургическое лечение до поступления ребенка в школу, прежде чем он/она будут психологически травмированы от жестоких насмешек сверстников.
При реконструкции ушной раковины при помощи силиконового имплантата хирургическое вмешательство можно проводить с 6-7 лет. При использовании реберного аутохряща операция должна быть отложена до тех пор, пока размер грудной клетки, и в частности хрящевой части ребер, будет достаточных размеров, чтобы обеспечить изготовление каркаса ушной раковины, что часто достигается в возрасте около 9-10 лет.
В исследованиях определено, что ушные раковины активно продолжают свой рост в среднем до 10 лет, это примерно на 85%, поэтому целесообразно проводить оперативное лечение в этот период, чтобы сформированное наружное ухо достаточно долго сохраняло симметрию с противоположным.
При этом отмечено, что реберный хрящ имеет большой потенциал к росту и сохраняет его при операциях формирования наружного уха. Поэтому пластически восстановленные из реберного хряща ушные раковины растут вместе с окружающими мягкими тканями в течение жизни пациента.
КАМУРАТИ-ЭНГЕЛЬМАННА БОЛЕЗНЬ
Камурати-Энгельманна болезнь (М. Camurati, современный итальянский врач; G. Engelmann, австрийский хирург и ортопед, род. в 1876 г.; син.: врожденные системные диафизарные гиперостозы, генерализованные системные гиперостозы с инволютивной миопатией, гиперпластический периостит, прогрессирующая диафизарная дисплазия, множественный диафизарный склероз, болезнь Энгельманна) — наследственный системный диафизарный гиперостоз у детей с инволютивной миопатией, относящийся к синдромам генерализованного остеосклероза. Чаще всего поражаются бедренные, плечевые и большеберцовые кости, в остальных костях изменения умеренные. Реже других поражаются передние и средние отделы основания черепа, лобные кости, тела позвонков, ногтевые фаланги.
Камурати-Энгельманна болезнь относят к редким семейным наследственным нарушениям развития костей. Предполагается доминантная передача мутантного гена. Описаны также спорадические случаи: Синглтон (Е. Singleton, 1956) наблюдал системное утолщение стенок сосудов, питающих кости, с сужением просвета артерий разного калибра, что дает основания приписать роль патогенетического фактора редуцированному кровоснабжению костной ткани.
Костная ткань в типичных случаях с выраженным остеосклерозом (см.), с сохранением общей структуры коркового и губчатого вещества, но резким сужением каналов остеона (гаверсовых каналов) и просветов сосудов. Костные балки, как правило, значительно утолщены.

Рис. 1. Больной системным гиперостозом: характерный вид длинных утолщенных бедер цилиндрической формы, саблевидных голеней, сгибательных контрактур в крупных суставах.
Ранний период жизни больного ребенка характеризуется замедленным развитием, небольшой прибавкой в весе, гипотрофией мышц конечностей. Ходить такие дети начинают на 3— 4-м году жизни. В дальнейшем наблюдается прогрессирующая мышечная слабость и развитие так наз. утиной походки. При тщательном осмотре можно обнаружить утолщение конечностей, приобретающих цилиндрическую форму без выпуклостей и углублений; суставы почти не выделяются (рисунок 1). Кожа над утолщенными костями натянута, отмечается уменьшение объема мышц (инволютивная Миопатия). Постоянным симптомом являются локализующиеся в диафизах больших трубчатых костей интенсивные, ноющие, «грызущие» боли, обычно связанные с периодом активации болезни и усиливающиеся при физ. нагрузке, тутоподвижность суставов и скованность движений. Описаны случаи непропорционального роста больных с выраженным удлинением конечностей, нарушением сухожильных рефлексов. Редко может развиться глухота и парез лицевого нерва по периферическому типу. Изменений крови и эндокринных органов обычно нет, однако М. В. Волков в 1974 г. сообщил о больной 19 лет, у к-рой были обнаружены инфантилизм, резкая гипоплазия гениталий и отсутствие менструаций, гипопластический субнанизм с отставанием умственного развития.
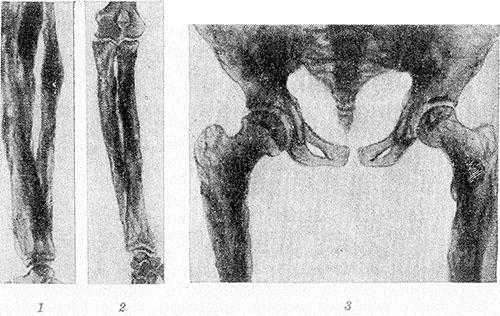
Рис. 2. Рентгенограммы костей голени (1), предплечья (2), таза и верхней трети бедер (3) больного системным гиперостозом: длинные трубчатые кости цилиндрически и булавовидно утолщены, корковый слой неплотный, резко утолщенный, имеет неравномерную пятнистую структуру с неровными контурами; кости таза утолщены.
Рентгенологически определяется симметричный гиперостоз трубчатых костей скелета. Пораженные кости громоздки, их диафизы утолщены в 2—3 раза, костно-мозговой канал равномерно сужен, но никогда не исчезает полностью. Эпифизы не изменены. Корковый слой значительно утолщен кнаружи и кнутри, с явлениями остеосклероза, но структура его не нарушена. Рисунок губчатого вещества изменен — костные балки грубы, толсты, плотны и обрывисты (рис. 2). В детском возрасте структурный рисунок может быть неравномерным, иметь рассеянный крупнопятнистый очаговый характер.
Камурати-Энгельманна болезнь следует дифференцировать с деформирующей остеодистрофией Педжета (см. Остоз деформирующий), акромегалией (см.), множественными кортикальными гиперостозами раннего возраста (см. Гиперостоз), болезнью Реклингхаузена (см. Нейрофиброматоз) и др. Важным дифференциально-диагностическим признаком, кроме типичной рентгенол, картины, отсутствия обезображивающих изменений, искривления костей и др., является отсутствие склонности к спонтанным переломам, несмотря на прогрессирование процесса.
Лечение симптоматическое. Назначают общеукрепляющие медикаменты и процедуры, при болях — анальгетики.
Течение заболевания медленное и доброкачественное. Прогноз для жизни благоприятный.
Библиография: Волков М. В. Болезни костей у детей, М., 1974; Каменева Т. И. и Трофимова 3. А. О врожденных системных, диафизарных гиперостозах у детей, Вопр. охр. мат. и дет., т. И, № 11, с. 89, 1966; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, кн. 1, с. 422, М., 1964; Ярандина М. П. Врожденный системный диафизарный гиперостоз — болезнь Энгельманна, Вестн, рентгенол, и радиол., № 3, с. 82, 1969; Camurati М. Symmetrical hereditary osteitis, Chir. Organi Mov., v. 6, p. 662, 1922; Engelmann G. Ein Fall von Osteopathia hyperostotica (Sclerotisans) multiplex infantilis, Fortschr. Rontgenstr., Bd 39, S. 1101, 1929.
Микротия
Микротия - это врожденное недоразвитие ушной раковины, сопровождающееся уменьшением ее размеров, деформацией или полным отсутствием. Клинически проявляется дисплазией завитка, противозавитка, мочки уха и устья слухового канала. Зачастую патология сочетается с пороками развития костей лицевого скелета. Диагностика заболевания основывается на результатах объективного осмотра, речевого исследования слуха, аудиометрии или импандансометрии, КТ и МРТ. Лечение микротии хирургическое, заключается в коррекции формы наружного уха, восстановлении проходимости ушного канала.
МКБ-10

Общие сведения
Микротия - редкая патология. Общая распространенность среди представителей европеоидной расы колеблется в пределах 1 на 8 500 - 10 000 новорожденных. В подавляющем большинстве случаев заболевание носит спорадический характер, менее чем у 18% больных детей удается установить наследственную связь. От 78% до 94% пациентов с этим пороком развития имеют поражение только одной ушной раковины, в 65-75% - правой. Представители мужского пола страдают в 1,5-2,5 раза чаще. Примерно у 35% больных наблюдается сопутствующее недоразвитие лицевого скелета, преимущественно нижней челюсти, канала лицевого нерва.

Причины микротии
Основная причина дизэмбриогенеза структур наружного уха - негативное внешнее влияние на развитие плода во время беременности. Наследственная микротия может являться составляющей одного из генетических синдромов - Нагера, Тричера-Коллинза, Конигсмарка, Гольденхара. К наиболее распространенным тератогенным факторам, обуславливающим развитие патологии, относятся:
- TORCH-инфекции. Это общее название инфекционных заболеваний, при которых имеется риск внутриутробного заражения плода и формирования пороков развития. Включают токсоплазмоз, герпес-вирусы типов 1, 2 и 3, краснуху, сифилис, цитомегаловирус, парвовирус.
- Физические факторы. Обычно это ионизирующее излучение при рентгенографии или компьютерной томографии, которые проводятся по жизненным показаниям или при недиагностированной беременности. Также в эту группу входят лучевая терапия при онкопатологиях, лечение радиоактивным йодом, продолжительная гипертермия.
- Вредные привычки. Губительное влияние на внутриутробное развитие ребенка оказывает употребление беременной алкогольных напитков, наркотических веществ (чаще всего - кокаина), табачных изделий.
- Фармакологические препараты. Некоторые медикаменты способны провоцировать врожденные аномалии у ребенка. Это антибиотики (тетрациклины, пеницилламин), гипотензивные (эналаприл, каптоприл) средства, препараты на основе йода или лития, антикоагулянты (варфарин), гормональные средства (андрогены).
- Эндокринные патологии. К ним относятся сахарный диабет в стадии декомпенсации, фенилкетонурия, недостаточность фолиевой кислоты, эндемический зоб. Отдельно выделяют гормонально активные опухоли, в том числе - андрогенпродуцирующие.
Патогенез
При нормальном эмбриональном развитии ткани будущей ушной раковины образуются из мезенхимы, окружающей первый эктодермальный карман - из I и II жаберных дуг. Первые зачатки ушных хрящей возникают на 20-22 неделе внутриутробного развития. Сенсорная часть слухового аппарата формируется раньше, из-за чего звуковоспринимающие структуры поражаются значительно реже. К 27-28 неделе раковины уже имеют внешний вид и форму как у новорожденного ребенка. Считается, что нарушение процесса развития вследствие влияния тератогенов может произойти в любом из вышеупомянутых периодов. Прослеживается закономерность: чем раньше подействовал негативный фактор - тем тяжелее будущие дефекты. Аномалии, возникшие до 6 акушерской недели, часто сопровождаются серьезными пороками или тотальным отсутствием не только наружного, но и среднего уха.
Классификация
В современной отоларингологии используется классификация Маркса, практическая значимость которой заключается в упрощении выбора хирургической тактики и метода слухопротезирования для пациентов. С учетом выраженности деформации раковины наружного уха и сопутствующего поражения слухового канала в ней выделяют 4 степени тяжести. Согласно этой классификации микротия может иметь следующие степени:
- I(легкая). Проявляется недоразвитием (гипоплазией) отдельных элементов ушной раковины. Слуховой проход сохраняет физиологическую форму или несколько сужается и полностью выполняет свои функции.
- II(средняя). Характеризуется значительной деформацией наружного уха с отсутствием некоторых элементов. Возникает стеноз или атрезия слухового прохода, сопровождающиеся тугоухостью по кондуктивному типу.
- III(тяжелая). Ушная раковина сильно недоразвита, представляет собой маленький рудимент. Просвет слухового канала, барабанная перепонка полностью отсутствуют.
- IV(анотия). Полное отсутствие раковины. Наблюдается агенезия структур наружного, среднего уха, нарушение формирования лицевого скелета.
Симптомы микротии
На основании морфологических характеристик выделяют 4 клинических формы заболевания: истинная микротия, анотия, малая и сложенная ушная раковина. Наиболее распространенная - истинная. Она проявляется наличием вертикального кожно-хрящевого валика с мочкой на конце. Последняя по сравнению с физиологическим расположением смещена кверху и кпереди. Слуховой проход часто заращен или отсутствует. Кости лицевого черепа, как правило, не деформированы. Анотия является самой редкой формой микротии. Внешне она характеризуется полным отсутствием ушной раковины или хрящевым бугорком, не имеющим мочки. Может сохраняться входное отверстие слухового канала. Часто сопровождается аномалиями развития лицевого скелета.
Сложенная раковина наружного уха представляет собой вариант микротии, при котором наблюдается недостаточное развитие верхней половины органа. Клинически это проявляется сращиванием ножки завитка и козелка. Из-за этого визуально ухо выглядит «свернутым», а его вертикальный размер существенно уменьшается. Противозавиток часто недоразвит. В тяжелых случаях верхний край завитка находится на одном уровне с козелком. Вся ушная раковина смещается кпереди и книзу. Сочетание этих изменений с дисплазией лицевого скелета является синдромом I жаберной дуги. Просвет слухового прохода зачастую сращен, слух на стороне поражения резко снижен или полностью отсутствует. Малая ушная раковина - разновидность заболевания, при которой имеется относительно сформированный завиток и мочка, недостаточно развитые противозавиток и углубления наружного уха, слуховой канал стенозирован или отсутствует.
Осложнения
Осложнения микротии связаны с несвоевременной коррекцией имеющихся дефектов слухового канала, возникающими на этой почве нарушениями слуха и речи. Тяжелая врожденная двухсторонняя кондуктивная тугоухость препятствует нормальному развитию артикуляционного аппарата у ребенка, становится причиной глухонемоты. Сужение входящего отверстия наружного уха нарушает дренаж отшелушившегося рогового эпителия и ушной серы. Это способствует размножению патогенной и условно-патогенной микрофлоры, приводит к наружным и средним отитам, мирингитам, мастоидитам, артритам височно-нижнечелюстных суставов и другим воспалительным поражениям этой области.
Диагностика
Постановка диагноза возможна сразу после родов на основании визуального осмотра. В последующем ребенок направляется к детскому отоларингологу для выявления сопутствующих поражений среднего и внутреннего уха, оценки функциональных возможностей звукопроводящей и звуковоспринимающей систем. Структура диагностической программы зависит от возраста пациента на момент обращения к специалисту. Помимо внешнего осмотра деформированной ушной раковины и сбора анамнеза она может включать в себя:
- Изучение слухового восприятия. Основывается на разговоре или использовании звучащих игрушек. Этот тест позволяет провести первичную оценку общего звукового восприятия и звукопроводимости каждого уха в отдельности. У младенцев вместо речи используются громкие внезапные звуки.
- Тональную пороговую аудиометрию. Дает возможность оценить воздушную проводимость и костное восприятие со стороны поражения. При микротии легкой, средней и тяжелой степени наблюдается кондуктивная тугоухость до 60-70 дБ. При анотии может присутствовать поражение звуковоспринимающего аппарата. Аудиометрия применяется у пациентов в возрасте от 3-4 лет, поскольку исследование требует адекватного восприятия звуковых сигналов и понимания сути теста.
- Акустическую импедансометрию. Используется для оценки функционального состояния барабанной перепонки, адекватности реакции цепи слуховых косточек на звук, определения патологий внутреннего уха. На основе полученных результатов устанавливается целесообразность слухопротезирования. При необходимости это методика дополняется ABR-тестом, изучающим реакцию ЦНС на звук.
- Компьютерную и магнитно-резонансную томографию.КТ височной кости в аксиальной, фронтальной и коронарной проекциях позволяет послойно визуализировать просвет наружного уха, барабанную полость и слуховые косточки или определить их отсутствие. У детей с рудиментарным проходом наружного уха по результатам исследования принимается решение о характере и объеме будущей операции, исключается наличие холестеатомы. Для диагностики потенциальных аномалий мягких тканей и изучения хода лицевого нерва показана МРТ височной кости.
Лечение микротии
Цели лечения - устранение косметического дефекта, улучшение слуховой функции, профилактика развития осложнений. Основной метод их достижения - хирургический. Выбор оперативного вмешательства зависит от выраженности микротии, степени развития слухового канала и сопутствующей дисплазии региональных костных структур. Из социальных и психологических соображений лечение рекомендуется проводить в дошкольном периоде - в 5-6 лет. В детской отоларингологии с этой целью используются:
- Аурикулопластика. В зависимости от клинической ситуации проводится пластика ушной раковины собственными тканями или (при III-IV ст.) с помощью имплантатов. Во втором случае используется аутотрансплантат, взятый из хряща VI, VII, VIII ребра с противоположной поражению стороны. Самая распространенная методика - многоэтапная реконструкция по Танзеру-Бренту.
- Меатотимпанопластика. Заключается в формировании косметически и функционально приемлемого слухового прохода при наличии такой возможности. В настоящее время обычно осуществляется меатотимпанопластика по методике С. Н. Лапченко.
- Слухопротезирование. При двухсторонней тяжелой кондуктивной тугоухости применяются слуховые аппараты с костным вибратором. При сохранении слухового прохода показаны классические слуховые проборы или кохлеарные имплантаты.
Прогноз и профилактика
Прогноз для здоровья пациента и косметический результат зависят от степени тяжести заболевания и возраста проведения лечебных мероприятий. Нарушение слуховой функции в большинстве случаев удается частично или полностью компенсировать. Специфических профилактических мер в отношении микротии не разработано. Неспецифическая профилактика подразумевает рациональное планирование беременности, ограничение или минимизацию влияния тератогенных факторов на плод: прием медикаментозных средств во время вынашивания ребенка строго по предписанию специалистов, отказ от вредных привычек, раннюю диагностику и лечение инфекционных патологий, эндокринных нарушений.
Аномалии развития ушной раковины
Аномалии развития ушной раковины - это группа врожденных патологий, которые характеризуются деформацией, недоразвитием либо отсутствием всей раковины или ее частей. Клинически может проявляться анотией, микротией, гипоплазией средней или верхней трети хряща наружного уха, в том числе свернутым или сросшимся ухом, лопоухостью, расщеплением мочки и специфическими аномалиями: «ухо сатира», «ухо макаки», «ухо Вильдермута». Диагностика основывается на данных анамнеза, объективного осмотра, оценки восприятия звука, аудиометрии, импедансометрии или ABR-теста, компьютерной томографии. Лечение хирургическое.



Аномалии развития ушной раковины - относительно редкая группа патологий. Согласно статистическим данным, их частота в различных частях планеты находится в пределах от 0,5 до 5,4 на 10 000 новорожденных. Среди лиц европеоидной расы показатель распространенности составляет 1 на 7 000 - 15 000 младенцев. Более чем в 80% случаев нарушения имеют спорадический характер. У 75-93% пациентов поражено только 1 ухо, из них в 2/3 случаев - правое. Примерно у трети больных пороки развития ушной раковины сочетаются с костными дефектами лицевого скелета. У мальчиков подобные аномалии встречаются в 1,3-2,6 раз чаще, чем у девочек.
Причины
Дефекты наружного уха - результат нарушений внутриутробного развития плода. Наследственные пороки встречаются относительно редко и входят в состав генетически обусловленных синдромов: Нагера, Тричера-Коллинза, Конигсмарка, Голденхара. Значительная часть аномалий формирования раковины уха обусловлена влиянием тератогенных факторов. Заболевание провоцируют:
- Внутриутробные инфекции. Включают инфекционные патологии из TORCH-группы, возбудители которых способны проникать через гематоплацентарный барьер. В этот список входят цитомегаловирус, парвовирус, бледная трепонема, рубелла, вирус краснухи, 1, 2 и 3-й типы герпес-вируса, токсоплазма.
- Физические тератогены. Врожденные аномалии ушной раковины потенцирует ионизирующее излучение при проведении рентгенологических исследований, длительное пребывание в условиях высоких температур (гипертермия). Реже в роли этиологического фактора выступает лучевая терапия при раковых заболеваниях, радиоактивный йод.
- Вредные привычки матери. Относительно часто нарушение внутриутробного развитие ребенка провоцирует хроническая алкогольная интоксикация, наркотические вещества, употребление сигарет и других табачных изделий. Среди наркотиков наиболее значимую роль играет кокаин.
- Медикаментозные средства. Побочным эффектом некоторых групп фармакологических препаратов является нарушение эмбриогенеза. К таким медикаментам относятся антибиотики из групп тетрациклинов, антигипертензивные, лекарства на основе йода и лития, антикоагулянты и гормональные средства.
- Заболевания матери. Аномалии формирования ушной раковины могут быть обусловлены нарушениями обмена веществ и работы желез внутренней секреции матери в период беременности. В перечень входят следующие патологии: декомпенсированный сахарный диабет, фенилкетонурия, поражения щитовидной железы, гормонопродуцирующие опухоли.
В основе формирования аномалий раковины уха лежит нарушение нормального эмбрионального развития мезенхимальной ткани, расположенной вокруг эктодермального кармана - I и II жаберной дуги. В нормальных условиях ткани-предшественники наружного уха образуются к концу 7 недели внутриутробного развития. На 28 акушерской неделе внешний вид наружного уха соответствует таковому у новорожденного ребенка.
Влияние тератогенных факторов в этот временной промежуток является причиной врожденных дефектов хряща ушной раковины. Чем раньше было оказано негативное воздействие - тем тяжелее его последствия. Более поздние повреждения не влияют на эмбриогенез слуховой системы. Воздействие тератогенов на сроке до 6 недель сопровождается тяжелыми пороками или полным отсутствием раковины и наружной части слухового прохода.
В клинической отоларингологии применяют классификации, основывающиеся на клинических, морфологических изменениях ушной раковины и прилегающих к ней структур. Основные цели деления патологии на группы - упрощение оценки функциональных возможностей пациента, выбора тактики лечения, решения вопроса о необходимости и целесообразности слухопротезирования. Широко используется классификация Р. Танзера, которая включает в себя 5 степеней выраженности аномалий ушной раковины:
- I - анотия. Представляет собой тотальное отсутствие тканей раковины наружного уха. Как правило, сопровождается атрезией слухового канала.
- II - микротия или полная гипоплазия. Ушная раковина присутствует, однако сильно недоразвита, деформирована или не имеет определенных частей. Выделяют 2 основных варианта:
- Вариант А - комбинация микротии с полной атрезией канала наружного уха.
- Вариант В - микротия, при которой слуховой проход сохранен.
- III - гипоплазия средней трети ушной раковины. Характеризуется недоразвитием анатомических структур, расположенных в средней части хряща уха.
- IV - недоразвитие верхней части ушной раковины. Морфологически представлена тремя подтипами:
- Подтип А - свернутое ухо. Наблюдается перегиб завитка вперед и вниз.
- Подтип В - вросшее ухо. Проявляется сращением верхней части задней поверхности раковины с кожей головы.
- Подтип С - тотальная гипоплазия верхней трети раковины. Полностью отсутствуют верхние участки завитка, верхняя ножка противозавитка, треугольная и ладьевидная ямки.
- V - лопоухость. Вариант врожденной деформации, при котором отмечается увлечение угла прилежания ушной раковины к костям мозговой части черепа.
В классификацию не включены локальные дефекты определенных участков раковины - завитка и мочки уха. К ним относятся бугорок Дарвина, «ухо сатира», раздвоение или увеличение мочки. Также в нее не входит непропорциональное увеличение уха за счет хрящевой ткани - макротия. Отсутствие перечисленных вариантов в классификации связано с низкой распространенностью этих пороков по сравнению с вышеупомянутыми аномалиями.
Симптомы
Патологические изменения можно выявить уже в момент рождения ребенка в родовом зале. В зависимости от клинической формы симптомы имеют характерные отличия. Анотия проявляется агенезией раковины и отверстия слухового прохода - на их месте располагается бесформенный хрящевой бугорок. Эта форма часто комбинируется с пороками развития костей лицевого черепа, чаще всего - нижней челюсти. При микротии раковина представлена смещенным вперед и вверх вертикальным валиком, на нижнем конце которого имеется мочка. При различных подтипах слуховой проход может сохраняться или быть заращенным.
Гипоплазия середины ушной раковины сопровождается дефектами или недоразвитием ножки завитка, козелка, нижней ножки противозавитка, чаши. Аномалии развития верхней трети характеризуются «загибанием» верхнего края хряща кнаружи, его сращением с расположенными позади тканями теменной области. Реже верхняя часть раковины полностью отсутствует. Слуховой канал при этих формах обычно сохранен. При лопоухости наружное ухо практически полностью сформировано, однако контуры раковины и противозавитка сглажены, а угол между костями черепа и хрящом составляет более 30 градусов, из-за чего последняя несколько «оттопыривается» кнаружи.
Морфологические варианты дефектов мочки уха включают аномальное увеличение по сравнению со всей раковиной, ее полное отсутствие. При раздвоении образуется два или более лоскута, между которыми имеется небольшая борозда, заканчивающаяся на уровне нижнего края хряща. Также мочка может прирастать к расположенным сзади кожным покровам.
Аномалия развития завитка в виде бугорка Дарвина клинически проявляется небольшим образованием в верхнем углу раковины. При «ухе сатира» наблюдается заострение верхнего полюса в сочетании со сглаживанием завитка. При «ухе макаки» наружный край несколько увеличен, средняя часть завитка сглажена или полностью отсутствует. «Ухо Вильдермута» характеризуется ярко выраженным выступанием противозавитка над уровнем завитка.
Осложнения аномалий развития ушной раковины связаны с несвоевременно проведенной коррекцией деформаций слухового канала. Имеющаяся в таких случаях выраженная кондуктивная тугоухость в детском возрасте приводит к глухонемоте или выраженным приобретенным нарушениям артикуляционного аппарата. Косметические дефекты негативно влияют на социальную адаптацию ребенка, что в некоторых случаях становится причиной депрессий или других психических расстройств.
Стенозы просвета наружного уха ухудшают выведение отмерших клеток эпителия и ушной серы, что создает благоприятные условия для жизнедеятельности патогенных микроорганизмов. Как результат формируются рецидивирующие и хронические наружные и средние отиты, мирингиты, мастоидиты, другие бактериальные или грибковые поражения региональных структур.
Постановка диагноза любой патологии этой группы основывается на внешнем осмотре области уха. Вне зависимости от варианта аномалии ребенка направляют на консультацию к отоларингологу для исключения или подтверждения нарушений со стороны звукопроводящего или звуковоспринимающего аппарата. Диагностическая программа состоит из следующих исследований:
- Оценка слухового восприятия. Базовый метод диагностики. Проводится при помощи звучащих игрушек или речи, резких звуков. В ходе теста врач оценивает реакцию ребенка на звуковые раздражители разной интенсивности в целом и со стороны каждого уха.
- Тональная пороговая аудиометрия. Показана детям старше 3-4 лет, что обусловлено необходимостью понимания сути исследования. При изолированных поражениях наружного уха или их сочетании с патологиями слуховых косточек на аудиограмме отображается ухудшение звуковой проводимости при сохранении костной. При сопутствующих аномалиях кортиевого органа снижаются оба параметра.
- Акустическая импедансометрия и ABR-тест. Эти исследования могут проводиться в любом возрасте. Цель импедансометрии - изучить функциональные возможности барабанной перепонки, слуховых косточек и выявить нарушение работы звуковоспринимающего аппарата. При недостаточной информативности исследования дополнительно используется ABR-тест, суть которого заключается в оценке реакции структур ЦНС на звуковой раздражитель.
- КТ височной кости. Ее применение оправдано при подозрении на выраженные пороки развития височной кости с патологическими изменениями звукопроводящей системы, холестеатому. Компьютерная томография выполняется в трех плоскостях. Также по результатам этого исследования решается вопрос о целесообразности и объеме операции.
Лечение аномалий развития ушной раковины
Основной метод лечения - оперативный. Его цели - устранение косметических недостатков, компенсация кондуктивной тугоухости и предотвращение осложнений. Подбор техники и объема операции основывается на характере и выраженности дефекта, наличии сопутствующих патологий. Рекомендуемый возраст проведения вмешательства - 5-6 лет. К этому времени заканчивается формирование ушной раковины, а социальная интеграция еще не играет столь важной роли. В детской отоларингологии используются следующие хирургические методики:
- Отопластика. Восстановление естественной формы ушной раковины выполняется двумя основными способами - при помощи синтетических имплантатов или аутотрансплантата, взятого из хряща VI, VII или VIII ребра. Проводится операция по Танзеру-Бренту.
- Меатотимпанопластика. Суть вмешательства - восстановление проходимости слухового канала и косметическая коррекция его входного отверстия. Наиболее распространенная методика - по Лапченко.
- Слухопротезирование. Целесообразно при тяжелой тугоухости, двухстороннем поражении. Применяются классические протезы или кохлеарные имплантаты. При невозможности компенсировать кондуктивное нарушение слуха при помощи меатотимпанопластики используются приборы с костным вибратором.
Прогноз для здоровья и косметический результат зависят от степени выраженности дефекта и своевременности проведенного оперативного лечения. В большинстве случаев удается достичь удовлетворительного косметического эффекта, частично или полностью устранить кондуктивную тугоухость. Профилактика аномалий развития ушной раковины состоит из планирования беременности, консультации врача-генетика, рационального приема медикаментов, отказа от вредных привычек, предотвращения воздействия ионизирующего излучения во время беременности, своевременной диагностики и лечения заболеваний из группы TORCH-инфекций, эндокринопатий.
1. Врожденные пороки развития ушной раковины у детей/ Водяницкий В.Б.// Детская больница. - 2012. - №3
2. Клинический протокол по диагностике и лечению пациентов с врожденными дефектами и деформациями ушных раковин. - 2014.
3. Хирургическое лечение врождённых пороков и аномалий развития ушной раковины/ Саидов И.З.// Вестник Авиценны. - 2011.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
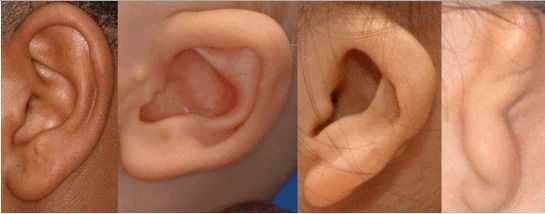
К лицевым аномалиям синдрома относятся:
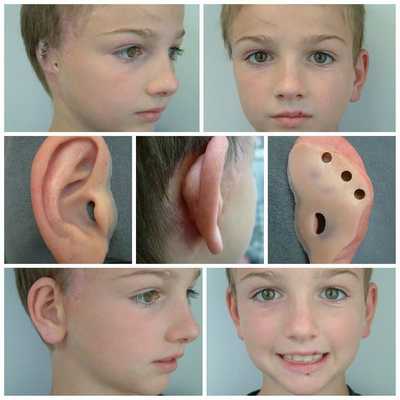
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
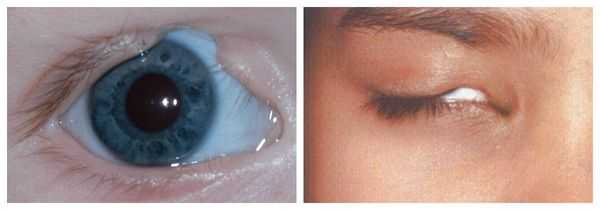
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
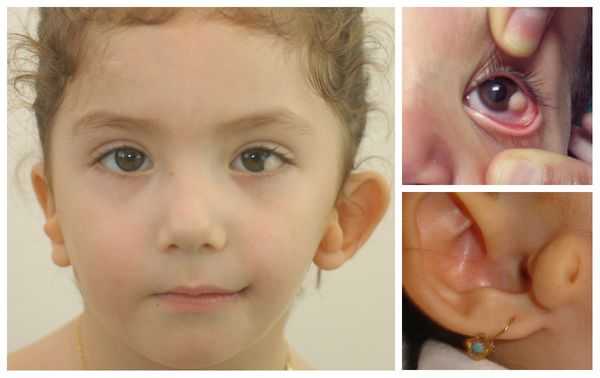
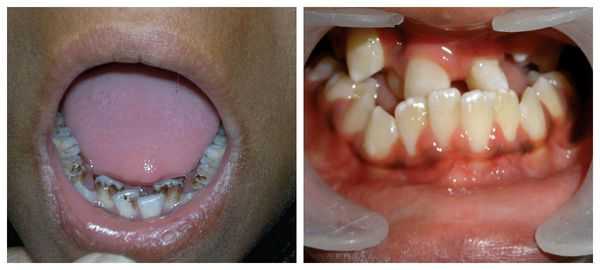
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
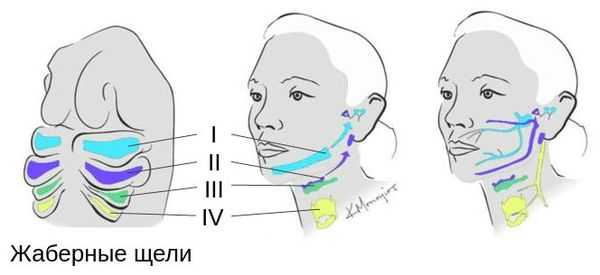
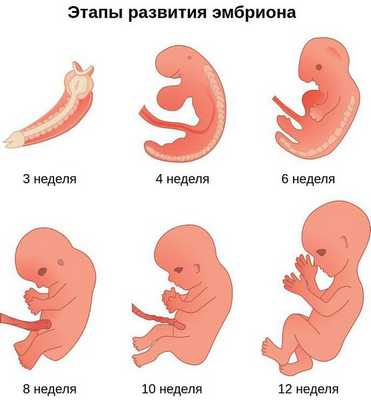
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
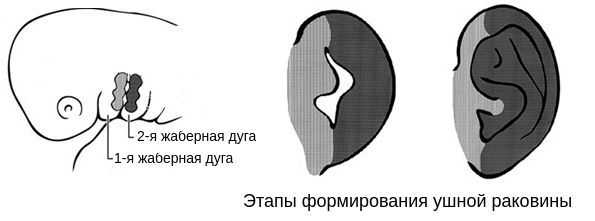
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
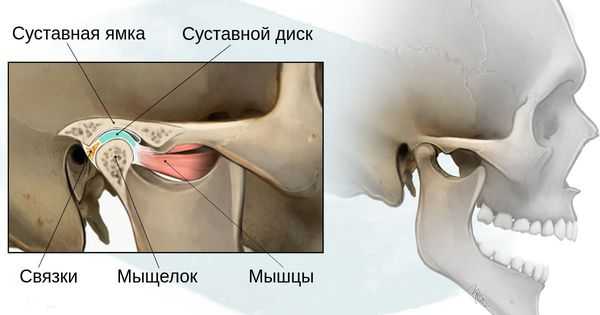
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Читайте также: