Глаза при синдроме MORM (mental retardation, truncal obesity, retinal dystrophy, micropenis)
Добавил пользователь Алексей Ф. Обновлено: 22.01.2026
Ювенильной макулярной дегенерацией или болезнью Штаргардта называют один из видов наследственной дистрофии макулярной области сетчатки. Заболевание выявляется в 12-20 летнем возрасте и проявляется прогрессирующим снижением остроты зрения обоих глаз.
В зависимости от местоположения патологии, принято выделять 4 формы ювенильной макулярной дегенерации:
- В области макулы;
- На средней периферии;
- В парацентральной области;
- В центральной и периферийной областях (смешанная форма).
Проводимые в настоящее время генетические исследования, доказывают, что ювенильная макулярная дегенерация и болезни Франческетти (жёлтопятнистое глазное дно), это фенотипические признаки одного заболевания.
Причины возникновения
Заболевание передается аутосомно-рецессивным путем наследования, редко аутосомно-доминантным. Посредством позиционного клонирования для ювенильной макулярной дегенерации определен основной локус вызывающего заболевание гена. Он экспрессируется в фоторецепторах и носит название АВСR. ABCR - это член суперсемейства т.н. АТФ-связывающего кассетного транспортёра, идентичный по последовательности человеческому гену RmP.
При аутосомно-доминантном пути наследования болезни, определена локализация генов мутации в хромосомах 13q, а также 6q14. Выявлено, что мутации ABCR присутствуют в субпопуляции больных с неэкссудативной формой макулодистрофии, связанной с возрастом и колбочко-палочковой дегенерацией, что позволяет предположить генетически обусловленный риск развития АМД у кровных родственников больных.
Видео нашего специалиста о заболевании

Симптомы заболевания
В пигментном эпителии сетчатой оболочки глаза возникает интенсивное накопление липофусцина. Процесс сопровождается ослаблением окислительной функции лизосом, с увеличением pH в клетках пигментного эпителия, что приводит к изменению их мембранной целостности.
При центральной форме ювенильной дистрофии, по мере развития заболевания, офтальмоскопическая картина области макулы имеет следующий вид: «битого металла», далее «бычьего глаза», затем «кованой бронзы» и в последствие - атрофии хороидеи.
Офтальмоскопия на стадии феномена «бычий глаз», выявляет тёмный центр, который окружён широким кольцом гипопигментации и, следующим за ним, еще одним кольцом гиперпигментации. Сосуды сетчатки - без изменений, ДЗН с темпоральной стороны бледен, что обусловлено атрофией нервных волокон папилломакулярного пучка. Фовеолярный рефлекс отсутствует, как и макулярное возвышение.
Наличие пятен желтовато-белого цвета, обнаруживается в ретинальном пигментном эпителии заднего полюса глаза. Пятна имеют различную величину, форму и конфигурацию — это наиболее характерный симптом желто-пятнистого глазного дна. Со временем форма, цвет и размеры пятен могут изменяться. Желтоватые первоначально пятна, с четко очерченными краями, спустя несколько лет нередко становятся серыми, границы их смазываются или исчезают.
Диагностика заболевания
В процессе сбора анамнеза, выясняется время возникновения заболевания (возраст манифестации), что играет важную роль в диагностике.
При лабораторных гистологических исследованиях в центральной области глазного дна отмечают увеличение пигмента, атрофию прилегающего пигментного эпителия сетчатки, комбинированную атрофию и гипертрофию пигментного эпителия. Представление желтых пятен липофусциноподобным материалом.
В ходе инструментальных исследований, при периметрии у пациентов с ювенильной макулярной дегенерацией обнаруживают разной величины относительные либо абсолютные центральные скотомы, что зависит от сроков процесса и его распространенности - с раннего детства или юношеского возраста. В случае желто-пятнистого глазного дна, изменений в области макулы не отмечается, поле зрения, зачастую, не изменено.
Цветоаномалии у большинства больных с центральной локализацией паталогического процесса развиваются по типу дейтеранопии или красно-зелёной дисхромазии, нередко более выраженной.
Цветовое зрение в случае желто-пятнистого глазного дна может не изменяться. Пространственная контрастная чувствительность значительно изменена во всех диапазонах пространственных частот, значительно снижена в зоне средних и полностью отсутствует в зоне высоких пространственных частот (т.н. паттерн-колбочковая дистрофия). Контрастная чувствительность отсутствует в центральной зоне сетчатки в границах 6-10 градусов.
Острота зрения, поле зрения и цветовое зрение находятся в норме. Темновая адаптация, как правило нормальная или незначительно снижена.
На ФАГ, в случае типичного феномена «бычьего глаза», при нормальном фоне выявляются зоны «отсутствия» или в ряде случаев гинофлюоресценции, с присутствием видимых хориокапилляров, а также «тёмная» или «молчащая» хороидея. Отсутствие в макулярной области флюоресценции объяснимо накоплением липофусцина, который экранирует флуоресцеин. Участки с гипофлюоресценцией иногда становятся гиперфлюоресцирующими в соответствии с зонами атрофии пигментно-эпителиального слоя.
Дифференциальная диагностика
Диагностику серьезно затрудняет сходство клинической картины многих дистрофических заболеваний в области макулы. Дифференциальный диагноз ювенильной макулярной дистрофии проводят с семейными друзами, пятнами сетчатки Кандори, прогрессирующей доминантной фовеальной дистрофией; ювенильным ретиношизисом; колбочковой, колбочко-палочковой, палочко-колбочковой дистрофиями; вителлиформной макулярной дистрофией; лекарственными дистрофиями.
Лечение и прогноз
Патогенетически обоснованного лечения ювенильной макулярной дегенерации до сегодняшнего дня нет. Необходимо постоянное наблюдение специалиста, контроль поля зрения, мониторинг ЭРГ, ЭОГ.
В зависимости от размера и выраженности поражения макулярной области, острота зрения прогрессивно снижается, особенно быстро, это происходит в детском и далее в юношеском возрасте.
Пациентам присваивается инвалидность с детства.
Профилактические рекомендации
В качестве меры профилактики, рекомендуется ношение солнцезащитных очков, что защитит от повреждающего макулу действия света, а также курсовой прием общеукрепляющих витаминов.
Обратившись в Московскую Глазную Клинику, каждый пациент может быть уверен, что за результаты хирургического вмешательства будут ответственны высококвалифицированные рефракционные хирурги - одни из лучших российских специалистов в данной области. Уверенности в правильном выборе, безусловно, прибавит высокая репутация клиники и тысячи благодарных пациентов. Самое современное оборудование для диагностики и лечения заболеваний глаз, одни из лучших специалистов и индивидуальный подход к проблемам каждого пациента - гарантия высоких результатов лечения в Московской Глазной Клинике.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
МКБ-10
Общие сведения
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу. Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет. В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Причины
Последние исследования генетического набора больных дистрофической миотонией показали, что основу заболевания составляет дефект в гене DMPK, находящемся в 19-й хромосоме и отвечающем за синтез миотонин-протеинкиназы. У больных дистрофической миотонией выявляется значительное увеличение тринуклеотидных CTG-повторов в основной части гена DMPK. При этом именно от количества повторов зависит форма и тяжесть миотонии.
В норме число тринуклеотидных повторов варьирует от 5 до 37. Увеличение повторов до 50-80 приводит к появлению мягкой формы миотонии Россолимо-Штейнерта-Куршмана. Если количество тринуклеотидных повторов находится в промежутке от 100 до 500, развивается поздняя форма заболевания. Врожденные формы дистрофической миотонии возникают при повышении числа CTG-повторов от 500 до 2000. Исследования показали, что увеличение тринуклеотидных повторов происходит в основном в женских гаметах в процессе мейоза. В связи с этим при передаче заболевания от матери у ребенка возникает более тяжелая форма миотонии или ее врожденный вариант.
Симптомы классической формы
В классическом варианте миотония Россолимо-Штейнерта-Куршмана начинает проявляться после первых 5 лет жизни и может манифестировать до 35-летнего возраста. Но наиболее часто клинические проявления заболевания возникают в возрастном диапазоне от 10 до 20 лет. Они представляют собой сочетание типичных симптомов миотонии с признаками миопатии, поражением сердечно-сосудистой системы и ЦНС, эндокринными нарушениями и катарактой.
Из миотонических проявлений для миотонии Россолимо-Штейнерта-Куршмана характерны миотонические спазмы, наиболее выраженные в жевательных мышцах и мышцах-сгибателях кисти. Наблюдаются также механические реакции миотонического типа, выявляемые при ударе неврологическим молоточком. Отличительной особенностью миотонии Россолимо-Штейнерта-Куршмана является наличие атрофических изменений в различных группах мышц. При этом течение заболевания характеризуется постепенным угасанием симптомов миотонии на фоне прогрессирующей мышечной дистрофии.
Чаще всего при миотонии Россолимо-Штейнерта-Куршмана поражаются мышцы дистальных отделов конечностей, мимическая мускулатура, грудино-ключично-сосцевидные и височные мышцы. Поражение мимических мышц проявляется характерным маскообразным печальным выражением лица больных дистрофической миотонией. Атрофические изменения мышц глотки и гортани приводят к развитию миопатического пареза гортани с нарушением голоса и затруднением глотания. Миопатические изменения могут возникать в дыхательной мускулатуре. Наряду с миотоническими спазмами они приводят к ухудшению легочной вентиляции, появлению приступов апноэ во сне, возникновению застойной или аспирационной пневмонии.
Нарушения сердечно-сосудистой системы наблюдаются примерно в половине случаев дистрофической миотонии. К ним относятся аритмии, связанные с нарушением проводимости, и гипертрофия левого желудочка. Наиболее распространена блокада ножек пучка Гиса. Из признаков поражения ЦНС чаще всего наблюдается гиперсомния и снижение интеллектуальных способностей, доходящее до легкой степени дебильности.
Эндокринные расстройства при миотонии Россолимо-Штейнерта-Куршмана затрагивают в основном половую сферу. У мужчин они проявляются снижением либидо, крипторхизмом, импотенцией, гипогонадизмом, у женщин — гирсутизмом, нарушениями менструального цикла (олигоменореей, дисменореей) и ранним климаксом. Типичным является изменение структуры волос в сочетании с алопецией. У мужчин отмечается выпадение волос на висках и в области лба, у женщин — диффузное или очаговое облысение.
Симптомы врожденной формы
Первые признаки врожденной формы миотонии Россолимо-Штейнерта-Куршмана могут проявляться еще в период внутриутробного развития. Как правило, они выражаются в значительном снижении двигательной активности плода, которое диагностируется акушером-гинекологом по данным акушерского УЗИ в III триместре беременности.
После рождения ребенка преобладают симптомы миопатии. Отмечается диффузная гипотония мышц, более выраженная в мимической, жевательной и глазодвигательной мускулатуре, а также в мышечных группах дистальных отделов конечностей. Характерны затруднения вскармливания и дыхательные расстройства. Миотоническая симптоматика начинает проявляться несколько позже. Врожденная дистрофическая миотония сопровождается задержкой моторного развития и олигофренией. Типично быстрое прогрессирование симптомов заболевания, часто приводящее к смертельному исходу еще в раннем детстве.
Диагностика
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологу предположить миотонию Россолимо-Штейнерта-Куршмана. Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ. К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии. Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия. От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
Радикальной терапии миотонии Россолимо-Штейнерта-Куршмана пока не существует. Пациентам, имеющим это заболевание, показана диета со сниженным содержанием калия. Им также следует избегать переохлаждения, которое провоцирует миотонические спазмы. Уменьшению миотонических проявлений способствует прием хинина, прокаинамида, фенитоина в сочетании с ацетазоламидом. Показаны анаболические стероиды ( нандролона деканоат, метиландростендиол, метандростенол), небольшие дозы АТФ, витамины группы В.
Синдром Мартина-Белл ( Синдром ломкой X-хромосомы )
Синдром Мартина-Белл - это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом - недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая - резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно - за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов - от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации - 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего - от 230 до 4 000.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем - 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм - увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания - умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания - своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм - полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка - неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ - 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще - мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Синдром Маркуса Гунна
Синдром Маркуса Гунна - это полиэтиологическое заболевание органа зрения, проявляющееся блефароптозом верхнего века, которое самопроизвольно приподнимается при движениях в височно-нижнечелюстном суставе. Диагностика включает в себя сбор анамнеза, проведение наружного осмотра, рентгенографии, магнитно-резонансной томографии (МРТ), офтальмоскопии, визометрии, биомикроскопии. Тактика лечения зависит от степени выраженности птоза. При минимальных проявлениях хирургическое вмешательство не показано. При тяжелом течении проводится тарзомиоэктомия, частичная резекция леватора верхнего века с его последующим укреплением при помощи аутотрансплантата.
Синдром Маркуса Гунна или пальпебромандибулярная синкинезия - это редкая, зачастую врожденная патология в офтальмологии, проявляющаяся птозом верхнего века в комбинации с синкинезиями. Данное заболевание впервые было описано в 1883 году английским офтальмологом М. Гунном, который наблюдал клиническую симптоматику синдрома у пятнадцатилетнего ребенка. Согласно статистическим данным, патология встречается у 5% больных с врожденным блефароптозом. Синдром Маркуса Гунна распространен повсеместно. Среди представителей женского и мужского пола встречается с одинаковой частотой. Как правило, первые признаки заболевания у детей выявляют в период грудного вскармливания. Родители обращают внимание на синхронные с открыванием рта движения верхнего века.
Врожденная форма синдрома Маркуса Гунна чаще развивается спорадически. В редких случаях отмечается аутосомно-доминантный тип наследования. Приобретенная форма может возникать у пациентов с прогрессирующей ишемией головного мозга, черепно-мозговой травмой, энцефалитом. Реже провоцирующим фактором выступает поражение лицевого нерва во время хирургических вмешательств, удаления зуба или инвазивных косметологических процедур. В механизме развития пальпебромандибулярной синкинезии Маркуса Гунна главную роль играет образование патологических связей между глазодвигательным, тройничным и лицевым нервами. При этом очаг аномальной импульсации расположен супрануклеарно.
Согласно филогенетической теории причиной развития заболевания является рудиментарная жаберно-оральная синкинезия, т. к. у рыб во время открытия ротовой полости жаберные дуги приподнимаются. Основой для их формирования у человека могут стать врожденные или приобретенные поражения пирамидной системы. При этом компенсаторная активизация подкорковых структур головного мозга провоцирует развитие клинических проявлений синдрома Маркуса Гунна.
Для синкинетического синдрома Маркуса Гунна характерен птоз верхнего века в сочетании с неконтролируемым подниманием века при движениях в височно-нижнечелюстном суставе. Первые проявления врожденной формы обнаруживаются во время кормления ребенка. При открытии рта пораженное блефароптозом веко поднимается. Данный процесс не контролируется волевыми усилиями и происходит при всех провоцирующих движениях нижней челюсти. В более зрелом возрасте больные отмечают, что открытие глаза также провоцируют движения нижней челюсти в сторону, противоположную зоне поражения, выдвижение кпереди, сжатие зубов и даже движения губами. Однако интенсивность клинической симптоматики может значительно варьировать у конкретных индивидуумов.
Как правило, синдром Маркуса Гунна развивается монокулярно, в редких случаях отмечается поражение обоих глаз. В раннем детском возрасте неспецифическими симптомами являются отечность века, слезотечение, общая слабость. Пациенты отмечают у себя уменьшение выраженности клинических проявлений заболевания с возрастом. В свою очередь, диагностические пробы указывают на то, что больные приобретают определенные навыки маскирования симптомов. Синдром Маркуса Гунна в большинстве случаев возникает совместно со страбизмом и амблиопией. Пальпебромандибулярная синкинезия может развиваться изолированно или сочетаться с синдромами Ваарденбурга и Гиршпрунга.
Диагностика синдрома Маркуса Гунна
Диагностика синдрома Маркуса Гунна основывается на анамнестических данных, результатах наружного осмотра, рентгенографии, магнитно-резонансной томографии (МРТ), офтальмоскопии. Дополнительные методы исследования - визометрия, биомикроскопия. В редких случаях в анамнезе у пациентов наблюдаются семейные формы заболевания, но чаще врожденная патология развивается спорадически. Больные приобретенной формой синдрома Маркуса Гунна зачастую отмечают травматические повреждения глаза, предшествующие появлению клинической симптоматики. При наружном осмотре визуализируется птоз одного, реже обоих век, который самостоятельно ликвидируется при движениях в височно-нижнечелюстном суставе.
Рентгенография проводится в случае приобретенной формы для выявления органических повреждений. Метод МРТ позволяет обнаружить патологическую взаимосвязь между глазодвигательным, тройничным и лицевым нервом. Как правило, аномальные очаги расположены супрануклеарно. Следствием указанных изменений может быть гипотрофия жевательной мышцы и круговой мышцы глаза. При проведении офтальмоскопии патологических изменений со стороны зрительного нерва не обнаруживается. Методом биомикроскопии можно выявить гиперемию и отечность конъюнктивы. Острота зрения, как правило, соответствует норме. Дифференциальную диагностику синдрома Маркуса Гунна необходимо проводить с врожденным птозом. При данной патологии движения нижней челюсти не способствуют поднятию века. Пальпебромандибулярная синкинезия относится к числу неврогенных птозов, поэтому для постановки диагноза помимо обследования у офтальмолога необходима консультация невропатолога.
Лечение синдрома Маркуса Гунна
Хирургическое лечение при синдроме Маркуса Гунна показано пациентам с выраженным птозом и синкинезиями, которые нарушают нормальную жизнедеятельность. Также показанием является пальпебромандибулярная синкинезия в сочетании со страбизмом и амблиопией. При легком течении заболевание не требует специфического лечения. Выбор методики операции зависит от степени выраженности птоза. В случае минимальных клинических проявлений рекомендовано проведение тарзомиоэктомии. При сохранении функциональной способности леватора верхнего века производят его частичную резекцию с последующим укреплением при помощи аутотрансплантата (связка Уитналла).
Для профилактики развития страбизма и амблиопии птоз у детей с тяжелым течением синдрома Маркуса Гунна устраняют после 3-4 лет. С целью ликвидации косметического дефекта у пациентов с неосложненным течением операцию рекомендовано проводить после 15 лет. Своевременные оперативные вмешательства обеспечивают восстановление функции век, нормальное развитие зрительного анализатора, но не могут полностью устранить проявления синкинеза. В послеоперационном периоде показано местное применение антисептических растворов для промывания, антибактериальных средств в виде капель или мазей.
Прогноз и профилактика синдрома Маркуса Гунна
Специфических методов профилактики синдрома Маркуса Гунна не разработано. Неспецифические превентивные меры приобретенной формы заключаются в соблюдении правил безопасности на производстве (ношение защитной маски, очков). При врожденном варианте патологии в детском возрасте рекомендовано носить пластырные повязки на время бодрствования. Это способствует нормальному развитию зрительного анализатора. Всем пациентам с установленным диагнозом необходимо 2 раза в год проходить осмотр у офтальмолога. Прогноз при синдроме Маркуса Гунна для жизни и трудоспособности благоприятный. Заболевание не приводит к снижению остроты зрения. В редких случаях возможно развитие вторичного конъюнктивита.
Ювенильная макулярная дегенерация (болезнь Штаргардта)
Ювенильная макулярная дегенерация сопровождается дистрофическими изменениями желтого пятна сетчатки. Первыми симптомами заболевания является двустороннее снижение зрения, возникающее в возрасте 10-20 лет.
Классификация
Выделяют несколько форм болезни Штаргардта, которые зависят от зоны распространения патологического процесса:
- Макулярная область;
- Средняя периферия;
- Парацентральная зона;
- Смешанная форма (патология расположена как в центре, так и на периферии).
Этиология
Путем генетического анализа было установлено, что ювенильная макулярная дегенерация наравне с желто-пятнистым глазным дном является фенотипическим проявлением одной и той же мутации. Тип наследования этой патологии обычно аутосомно-рецессивный, но иногда и аутосомно-доминантный.
Методом позиционного клонирования установлен локус гена, который экспрессируется в фоторецепторах.
При болезни Штаргардта происходит выраженное накопление липофусцина, которые ингибирует окислительную функцию лизосом. В результате увеличивается кислотность клеток глазного дна и нарушается их мембранная целостность.
Клиническая картина
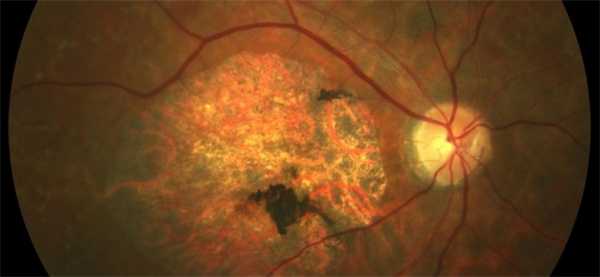
Если речь идет о центральной дистрофии Штаргардта, то внешне зона макулы выглядит как «бычий глаз», «битый металл», «кованая бронза» или атрофия хориоидеи.
При «бычьем глазе» имеется темная центральная зона, которая окружена кольцом гипопигментации. Далее обычно следует еще одно кольцо гиперпигментации. При этом сосуды сетчатки не изменены, отмечается бледность диска зрительного нерва с темпоральной стороны (атрофия нервных клеток в папилломакулярном пучке). Макулярное возвышение и фовеолярный рефлекс отсутствуют.
При желто-пятнистом глазном дне в заднем полюсе глаза (ретинальный пигментый эпителий) имеются желтовато-белые пятна, форма и величина которых различна. С течением времени величина и форма пятен изменяется, цвет из становится из желтого сероватым, а границы размываются.
Важную роль в диагностике ювенильной макулярной дегенерации играет время появления первых симптомов (в детском или юношеском возрасте).
При гистологическом исследовании можно обнаружить увеличение пигмента в центральной области глазного дна. Также возникает атрофия или комбинация атрофии и гипертрофии пигментного эпителия сетчатки. Вещество желтых пятен состоит из липофусциноподобного материала.
Периметрия у пациентов с заболеванием Штаргардта позволяет выявить относительные или абсолютные скотомы. Размер их зависит от сроков заболевания и его распространенности. Если речь идет о желто-пятнистом глазном дне, то макулярная зона обычно не задействована, поэтому изменений поля зрения может не быть.
Цветоаномалия чаще всего возникает при центральной локализации и проявляется дейтеранопией или красно-зеленой дисхромазией.
Иногда желто-пятнистое глазное дно не сопровождается снижением зрения. Однако пространственная контрастная чувствительность снижается во всех диапазонах частот (особенно в зоне средних пространственных частот). В центральной зоне в пределах 6-10 градусов отсуствует контрастная чувствительность колбочковой системы.
В начальных стадиях болезни Штаргардта центральной формы отмечается снижение макулярной электроретинограммы, а в поздних стадиях она отсутствует. Если поражается периферия, то изменения возникают только в развитых стадиях и проявляются снижением колбочковых и палочковых компонентов ретинограммы. При этом симптомы у пациентов, как правило, отсутствуют, а острота, поле зрения и цветовосприятие находятся в пределах нормы. Может быть незначительно снижена темновая адаптация.
При флуоресцентной ангиографии при нормальном фоне выявляют области гипофлуоресценции (или ее отсутствия) с видимыми хориокапиллярами. В макулярной области отсутствует свечение в результате накопления липофусцина, который экранирует флуоресцеин. Если зоны гипофлуоресценции становятся гиперфлуоресцирующими, то это говорит об атрофии пигментного эпителия сетчатки.
Диагностику может затруднять сходство клинических проявлений различного рода дистрофий макулы. Болезнь Штаргардта нужно отличать от семейных друз, пятен сетчатки Кандори, колбочковой (палочково-колбочковой) дистрофии, доминантной прогрессирующей дистрофии фовеа, ювенильного ретиношизиса, приобретенных лекарственных дистрофий, вителлиформной макулярной дистрофии.
Лечение
Провести патогенетически обоснованное лечение невозможно, поэтому пациенты с ювенильной макулярной дегенерацией являются инвалидами с детства. Эти пациенты требует мониторного наблюдения с определением границ поля зрения, выполнением электроретинографии и электроокулографии.
Чтобы уменьшить негативные последствия, рекомендуется пользоваться солнцезащитными очками и применять общеукрепляющие витаминные препараты.
Прогноз
При болезни Штаргардта отмечается прогрессивное снижение зрительной функции (особенно в юношеском или детском возрасте), что является следствием выраженных изменений в макуле.
Читайте также: