Легкие талассемические и талассемическоподобные синдромы: дельта- и гамма-талассемии, синдром Lepore
Добавил пользователь Валентин П. Обновлено: 21.01.2026
(средиземноморская анемия; большая и малая талассемия)
, MD, PhD, Johns Hopkins University School of Medicine
- Патофизиология
- Клинические проявления
- Диагностика
- Прогноз
- Лечение
- Основные положения
- Дополнительная информация
Талассемии - это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского, средиземноморского, или южноазиатского происхождения. Бета-талассемия более распространена у лиц средиземноморского, ближневосточного, южноазиатского и индийского происхождения. Симптомы и признаки обусловлены анемией, гемолизом, спленомегалией, гиперплазией костного мозга, при многократных гемотрансфузиях может наблюдаться перегрузка железом. Диагностика основана на генетическом исследовании и количественном анализе структуры гемоглобина. Лечение тяжелых форм может включать в себя гемотрансфузии, спленэктомию, терапии хелаторами и трансплантацию стволовых клеток.
Патофизиология талассемий
Талассемия - гемоглобинопатия Обзор гемоглобинопатий Гемоглобинопатии - это генетические нарушения, влияющие на структуру или синтез молекулы гемоглобина. Молекулы гемоглобина состоят из полипептидных цепей, химическая структура которых контролируется. Прочитайте дополнительные сведения , которая является одним из наиболее распространенных наследственных заболеваний, связанных с синтезом гемоглобина. Нормальная зрелая молекула гемоглобина (гемоглобин А) состоит из 2 пар цепей, называемых альфа и бета. Нормальная кровь взрослого человека также содержит ≤ 2,5% Hb A2 (состоит из альфа- и дельта-цепей) и 2% гемоглобина F (фетального гемоглобина), который имеет гамма-цепи вместо бета-цепей. Талассемия является результатом несбалансированного синтеза гемоглобина, вызванного снижением синтеза по крайней мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта).
Альфа-талассемия
Альфа-талассемия является результатом снижения синтеза альфа-полипептидных цепей вследствие делеции одного или нескольких генов альфа-цепей. Люди, как правило, имеют четыре гена альфа-цепей (по два на каждой паре хромосом), потому что ген альфа-цепей дублируется. Классификация болезни основана на количестве и местоположении делеций:
Альфа + талассемия: Потеря одного гена на одной хромосоме (альфа/--)
Альфа 0 талассемии: Потеря обоих генов на одной и той же хромосоме (--/--)
Бета-талассемия
Бета-талассемия вызвана снижением синтеза бета-полипептидных цепей в результате либо мутации, либо делеции в гене бета-глобина, что приводит к нарушению синтеза гемоглобина А. Мутации или делеции могут привести к частичной потере (бета + аллель) или полной потере (бета 0 аллель) функции бета-глобина. Существуют два гена бета-глобина, и у пациентов могут быть гетерозиготные, гомозиготные или сложные гетерозиготные мутации. Кроме того, пациенты могут быть гетерозиготными или гомозиготными по аномалиям в 2-х различных генах глобина (например, бета и дельта).
Бета-дельта-талассемия является менее распространенной формой бета-талассемии, при которой нарушается синтез как дельта-цепи, так и бета-цепи. Эти мутации могут быть гетерозиготными или гомозиготными.
Симптомы и признаки талассемий
Клинические особенности талассемий сходны, но различаются по степени тяжести, в зависимости от количества нормального гемоглобина.
Пациенты с одной альфа + аллелью (альфа/альфа; альфа/--) являются клинически нормальными и называются бессимптомными носителями.
У гетерозигот с дефектами в 2 из 4 генов, таких как две альфа + аллели (альфа/--; альфа/--) или 1 альфа 0 аллель (альфа/альфа; --/--) наблюдается тенденция к развитию микроцитарной анемии легкой или умеренной степени тяжести, но с субклиническим течением. Данные пациенты имеют малую альфа-талассемию.
Дефекты в 3 из 4 генов, вызванные совместным наследованием как альфа +, так и альфа 0 (альфа/-; -/-), существенно нарушают синтез альфа-цепи. Синтез поврежденных альфа-цепей в результате приводит к образованию тетрамеров избыточных бета-цепей, называемых Hb H, а у младенцев - к формированию гамма-цепей, называемых гемоглобином Барта. У пациентов с болезнью гемоглобина Н часто наблюдаются гемолитическая анемия и спленомегалия.
Дефект всех 4 генов через две альфа 0 аллели (--/--;--/--) является летальным состоянием, которое вызывает внутриутробную гибель плода (водянку плода), поскольку гемоглобин, не содержащий альфа-цепей, не переносит кислород.
При бета-талассемии, клинические фенотипы подразделяются на 3 группы в зависимости от степени нарушения синтеза бета-глобина:
Малая бета-талассемия (характерно) возникает у обычно бессимптомных гетерозигот (бета/бета + бета/бета 0) с клинической картиной микроцитарной анемии от легкой до умеренной степени. Этот фенотип может также возникнуть в легких случаях бета +/бета +.
Промежуточная бета-талассемия проявляется вариабельной клинической картиной, которая является промежуточной между большой и малой талассемией, обусловленная наследованием 2 аллелей бета-талассемии (бета +/бета 0 или, в тяжелых случаях, бета +/бета +).
Большая бета-талассемия (или анемия Кули) возникает у гомозиготных пациентов (бета 0/бета 0) или сложных гетерозигот (бета 0/бета +) в результате тяжелого дефекта бета-глобина. У этих пациентов развивается тяжелая анемия и гиперактивность костного мозга. Большая бета-талассемия проявляется в возрасте от 1 до 2 лет с симптомами тяжелой анемии и трансфузионной и абсорбционной перегрузки железа. У пациентов наблюдаются желтуха, язвы нижних конечностей и холелитиаз (как при серповидноклеточной анемии Серповидно-клеточная анемия Серповидноклеточная анемия ( гемоглобинопатия) является причиной хронической гемолитической анемии, которая наблюдается практически исключительно у представителей негроидной расы. Она вызвана. Прочитайте дополнительные сведенияДиагностика талассемий
При наличии подозрения диагностика данного типа гемолитической анемии включает в себя:
Мазок периферической крови
Исследование структуры ДНК (пренатальная диагностика)
Малую талассемии обычно выявляют при проведении рутинного мазка периферической крови и развернутого анализа крови, когда обнаруживают микроцитарную анемию и повышенное количество эритроцитов. При желании, диагноз малой бета-талассемии может быть подтвержден с помощью количественных исследований структуры гемоглобина. Никакого вмешательства не требуется; у женщин анемия может усугубляться беременностью.
Более тяжелую талассемию следует подозревать у пациентов с отягощенным наследственным анамнезом, при наличии характерных клинических симптомов или микроцитарной гемолитической анемии. При подозрении на талассемию выполняются обычные лабораторные тесты для выявления микроцитарных гемолитических анемий и количественный анализ структуры гемоглобина. Характерно повышение уровня билирубина, железа и ферритина в сыворотке крови.
При альфа-талассемиях процентное содержание Hb F и Hb A2 обычно в пределах нормы, выявление одного или двух дефектных генов, характерных для талассемии, производится с помощью современных генетических тестов. Диагноз часто ставится путём исключения других причин микроцитарной анемии.
При большой бета-талассемии наблюдается тяжелая анемия, часто со снижением уровня гемоглобина ≤ 6 г/дл (≤ 60 гр/л). Количество эритроцитов повышено по отношению к уровню гемоглобина, поскольку наблюдается выраженный микроцитоз. Диагностика основана на исследовании мазка периферической крови, в котором обнаруживается множество ядросодержащих эритробластов, мишеневидных клеток, небольших бледноокрашенных эритроцитов; характерна точечная или диффузная базофилия.
При количественном анализе структуры гемоглобина диагностическим критерием малой бета-талассемии является повышенный уровень Hb A2. При большой бета-талассемии обычно повышено содержание Hb F, иногда до 90%, а содержание Hb A2 обычно > 3%.
Болезнь гемоглобина Н диагностируется по выявлению Hb H или фракций Барта при электрофорезе гемоглобина. Наличие специфического молекулярного дефекта не меняет клинического подхода.
Стандартом пренатальной диагностики и генетического консультирования является картирование генов рекомбинантной ДНК (в частности метод полимеразной цепной реакции [ПЦР]).
Если при анемии выполняется исследование костного мозга (к примеру, для исключения других причин), выявляется выраженная эритроидная гиперплазия.
При визуальном исследовании, выполненном по другим причинам, у пациентов с большой бета-талассемией можно выявить изменения, обусловленные хронической гиперактивностью костного мозга. Наблюдаются истончение кортикального слоя костей черепа, расширение диплоических пространств, лучистая трабекулярная структура, появление гранул или феномен «матового стекла». В трубчатых костях могут наблюдаться очаги остеопороза, расширение костномозгового пространства и истончение кортикального слоя. Тела позвонков могут иметь зернистый внешний вид, или вид матового стекла. Фаланги могут быть прямоугольными или двояковыпуклыми. Визуализация грудной клетки может выявить признаки паравертебрального экстрамедуллярного гемопоэза.
Прогноз при талассемии
Продолжительность жизни у пациентов с малой бета-талассемией или малой альфа-талассемией является нормальной. Прогноз при заболевании Hb Н и промежуточной бета-талассемии вариабельный.
Продолжительность жизни снижена у пациентов с большой бета-талассемией, главным образом в связи с осложнениями в результате хронических трансфузий.
Лечение талассемий
Часто - переливание эритроцитарной массы с/без железохелатирующей терапии
Спленэктомия при наличии спленомегалии
Если возможно, выполняют трансплантацию аллогенных стволовых клеток
Луспатерцепт для лечения трансфузионно-зависимой бета-талассемии
Пациентам с малой альфа-талассемией или малой бета-талассемией лечение не требуется.
При гемоглобинозе Н: спленэктомия может помочь при тяжелой анемии или наличии спленомегалии.
Пациенты с промежуточной бета-талассемией должны получать как можно меньше гемотрансфузий, чтобы избежать перегрузки железом. Тем не менее супрессия патологического гемопоэза с помощью периодических трансфузий эритроцитов Эритроциты (ККТ) Цельная кровь способствует улучшению кислородной емкости крови, расширению объема и замене факторов свертывания, а раньше ее назначали при сильной кровопотере. Однако, поскольку компонентная. Прочитайте дополнительные сведения может быть ценной у пациентов с тяжелым течением заболевания. При большой бета-талассемии следует проводить трансфузии по необходимости для поддержания уровня гемоглобина примерно 9 - 10 г/дл (от 90 до 100 г/л) и избегать развития тяжелых клинических проявлений.
Чтобы предотвратить или отсрочить развитие осложнений от перегрузки железом, должен быть удален избыток (трансфузионного) железа (например, путем назначения длительной железохелаторной терапии Лечение Вторичная перегрузка железом появляется в результате избыточной абсорбции железа, повторяющихся переливаниях крови или избытке перорального приема, как правило, у пациентов с нарушениями эритропоэза. Прочитайте дополнительные сведения ). Хелаторную терапию, как правило, начинают, когда уровни сывороточного ферритина превышают 1000 нг/мл (> 1000 мкг/л) или после приблизительно от 1 до 2 лет проведения плановых трансфузий. Спленэктомия может снизить потребность в гемотрансфузиях у пациентов с выраженной спленомегалией.
Люспатерцепт представляет собой инъекционный рекомбинантный белок слияния, который ингибирует метаболический путь передачи сигналов от трансформирующего фактора роста бета. В рандомизированном плацебо-контролируемом исследовании у пациентов с бета-талассемией он снизил потребность в трансфузии на 33% у 21% пациентов (по сравнению с 4,5% в контрольной группе). Луспатерцепт является вариантом лечения для пациентов, которым необходимо переливание крови (1 Справочные материалы по лечению Талассемии - это группа врожденных микроцитарных гемолитических анемий, которые характеризуются дефектом синтеза гемоглобина. Альфа-талассемия особенно распространена среди лиц африканского. Прочитайте дополнительные сведения ).
Справочные материалы по лечению
1. Cappellini MD, Viprakasat V, Taher A, et al: A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med 382(13):1219-1231, 2020. doi: 10.1056/NEJMoa1910182
Основные положения
Талассемия является результатом снижения синтеза по меньшей мере одной полипептидной цепи глобина (бета, альфа, гамма, дельта); в результате аномальные эритроциты являются микроцитарными, часто неправильной формы и склонны к гемолизу (вызывающему анемию).
Часто проявляется спленомегалия, часто массивная, которая может привести к селезеночной секвестрации, ускоряющей разрушение эритроцитов (в том числе введенных при переливании).
Часто наблюдается перегрузка железом из-за увеличенного поглощения (в связи с нарушенным эритропоэзом), а также из-за частых трансфузий.
Диагностика с использованием электрофореза гемоглобина.
Следует проводить трансфузии по необходимости, проводя мониторинг перегрузки железом и используя хелаторную терапию.
Спленэктомия может снизить потребность в гемотрансфузиях у пациентов со спленомегалией.
Трансплантация аллогенных стволовых клеток имеет излечивающий эффект.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Cooley's Anemia Foundation: provides comprehensive patient education and support and advocacy to patients with thalassemia
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Талассемия — болезнь, вызванная гемоглобином

Талассемия — это не одна, а группа похожих болезней, передающихся по наследству и вызванных нарушенным синтезом белковой части гемоглобина. Заболевание достается ребенку с вероятностью в 100%, если один из родителей содержит измененный ген. Поэтому оно считается самой распространенной наследственной патологией.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Почему разрушаются эритроциты?
Патогенез талассемии достаточно изучен. Первичные изменения начинаются с нарушенного синтеза гемоглобина, а именно белковых цепочек вещества. Известно, что гемоглобин, входящий в состав 90% массы эритроцитов, — единственное вещество, способное связывать молекулы кислорода и разносить их из легочной ткани по всему организму.
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.
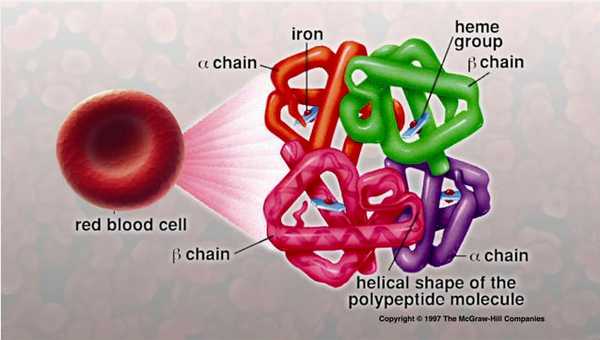
Основные четыре цепочки белков, связывающие гем и влияющие на эритроциты
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
- «немой» ген (α-th2);
- «манифестный» ген (α-th1).
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Клинические проявления большой талассемии
Симптомы талассемии зависят по клинике и времени проявления от вида генной мутации.
Большая талассемия (болезнь Кули) развивается при гомозиготной передаче от родителей. Ее проявления заметны у детей сразу после рождения.
У новорожденного замечают:
- удлиненный вверх череп («башенный»);
- более развитую верхнюю челюсть;
- монголоидный тип лицевого скелета.
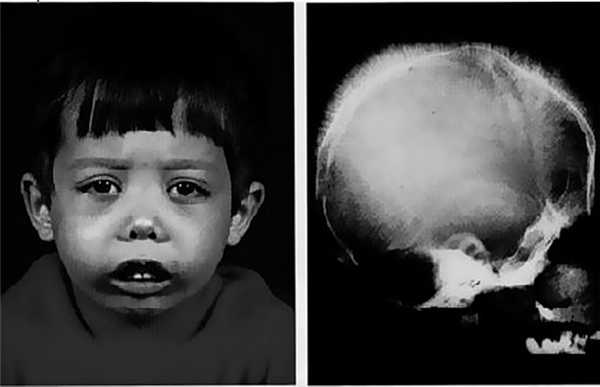
Типичная внешность и изменения структуры костей черепа используются в диагностике
К 12-месячному возрасту проявляется:
- расширенная носовая перегородка, «приплюснутый» нос;
- костные наросты на стопах;
- нарушенный прикус;
- желтушность кожи (в связи с поражением селезенки).
Кислородное голодание тканей приводит к органным нарушениям. При осмотре отмечаются:
- увеличение печени, край плотный на ощупь (развивается ранний цирроз);
- возникновение сердечной недостаточности (лишнее железо откладывается в миокарде и поражает сократимость сердечной мышцы);
- отставание в физическом и умственном развитии от сверстников.
У малыша развивается сахарный диабет из-за фиброзирования поджелудочной железы и печеночная недостаточность. В тяжелых случаях он живет не более одного года.
В старшем возрасте проявляются такие осложнения:
- трофические язвы на коже, вызванные локальными нарушениями кровообращения;
- частые переломы костей;
- повторные воспаления в легких;
- калькулезный холецистит;
- кардиосклероз;
- сепсис при контакте с инфекцией;
- нарушенное полового развитие.
Выделяют промежуточную форму β-талассемии с более доброкачественным течением. Она начинается у детей старшего возраста. Внешний вид ребенка не изменяется. Нет отставания в умственном и физическом развитии. Основные проявления вызываются увеличением селезенки и повышенной ломкостью костной ткани.
Клиника малой талассемии
Малая или гетерозиготная форма передается одним из родителей. Второй «здоровый» ген сглаживает повреждения. Симптомы талассемии могут длительно не проявляться или вызывать признаки, общие с другими заболеваниями:
- слабость, повышенную утомляемость;
- головные боли;
- головокружение.
При осмотре обращается внимание на следующее:
- бледность кожи с желтушным оттенком;
- возможно увеличение печени и селезенки.
У ребенка с малой формой резко увеличена восприимчивость к инфекциям, снижен иммунитет. Очень тяжело и часто протекают вирусные и кишечные инфекционные заболевания.
Во взрослом состоянии у женщины при беременности развивается водянка плода (повышение внутричерепного давления и скопление жидкости в желудочках мозга). Эта патология несовместима с жизнью ребенка, в случае нормальных родов ведет к тяжелым неврологическим и психическим отклонениям.
Минимальной β-талассемией называют синдром Сильвестрони — Бьянко. Эта форма заболевания протекает практически бессимптомно, обнаруживается случайно в семьях со случаями талассемии.
Диагностика
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
В анализе крови выявляют:
- снижение уровня гемоглобина до 30-50 г/л при гомозиготном варианте и до 90 -110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
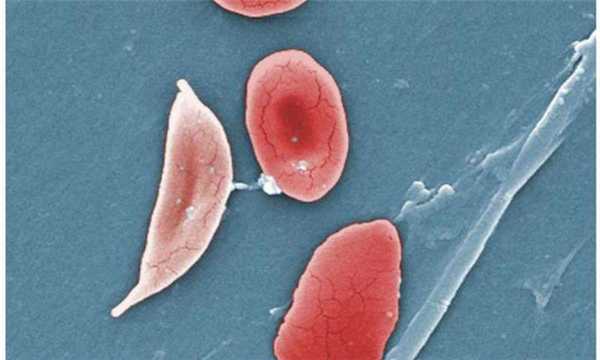
Дрепаноциты образуются при любых гемоглобинопатиях, содержат особую разновидность (S гемоглобин), который может в условиях низкой концентрации образовывать полимеры и изменять форму оболочки клетки
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Пересадка костного мозга считается наиболее результативным способом
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8-10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.
Можно ли предупредить заболевание?
Талассемия считается до настоящего времени неизлечимым заболеванием. К мерам предупреждения относят дородовую диагностику для решения вопроса о наследовании признаков, своевременного прерывания беременности.
В родильном доме берется кровь из пяточки для экспресс-теста
Рожденный ребенок с легкой степенью болезни может прожить всю жизнь без тяжелых последствий. Но гарантировать именно эту форму патологии современная наука не может. Особенно следует подумать о целесообразности деторождения парам с гомозиготной наследственностью.
Методика исследования клеток плода во время беременности (забор осуществляется путем прокола матки через брюшную стенку) опасна возможным занесением инфекции и преждевременными родами. Поэтому лучше решать проблемы в медико-генетической консультации до плановой беременности.
Больных с талассемией ведут и наблюдают совместно гематологи, педиатры и терапевты.
Таламический синдром ( Синдром Дежерина-Русси , Синдром таламической боли )
Таламический синдром - это центральный болевой синдром, спровоцированный повреждением таламуса. Непосредственной причиной заболевания выступает сосудистое поражение, которое вызывает инфаркт вентропостериомедиальных и вентропостериолатеральных таламических ядер. Патология проявляется сильными болями и нарушениями чувствительности на стороне, противоположной зоне поражения в головном мозге. Диагностика включает оценку неврологического статуса, инструментальную нейровизуализацию, консультацию психиатра. Для купирования таламических болей назначают психотропные препараты и опиоидные анальгетики, в рефрактерных случаях проводится таламотомия.
МКБ-10



Общие сведения
Таламический синдром (синдром Дежерина-Русси) - распространенный вариант хронической боли, который развивается у 8% пациентов после острых нарушений мозгового кровообращения. Клиническую картину заболевания впервые описали в 1906 г. французские врачи: невролог Ж. Дежерин и патолог Г. Русси, которые изучали симптомы у больных после инсульта. Патология имеет большое значение в практической неврологии ввиду своей частой встречаемости, многообразных и мучительных для пациента симптомов, сложностей при подборе эффективной схемы фармакокоррекции.

Причины
Основной этиологический фактор таламического синдрома - ишемические и геморрагические инсульты. Большинство случаев заболевания вызваны повреждением вентропостериолатерального отдела таламуса. Реже симптоматика возникает при патологии вентропостериомедиальных ядер. Непосредственной патоморфологической причиной клинических проявлений признан тромбоз таламо-коленчатой артерии, ветви задней мозговой артерии.
Среди факторов риска на первое место ставят гипертоническую болезнь, которая вызывает до 80% всех кровоизлияний в головной мозг. При ишемической форме инсульта основным провоцирующим фактором является атеросклероз церебральных и экстракраниальных сосудов. Повышенная вероятность поражения таламуса и формирования таламического синдрома наблюдается у пациентов с болезнями сосудов: системными васкулитами, амилоидозом, артериовенозными мальформациями. Независимым фактором риска выступают гемофилии, тромбоцитопении и другие нарушения свертывания крови.
Патогенез
Таламус (зрительный бугор) - часть промежуточного мозга, которая имеет яйцевидную форму и объем около 3 см 3 . Он содержит более 40 ядер — скоплений тел нейронов, которые отделены друг от друга прослойками белого вещества. По локализации выделяют переднюю, центральную, медиальную, латеральную и заднюю группу таламических ядер. Орган выполняет сенсорную, моторную и ассоциативную функции.
Заднелатеральное ядро, которое чаще всего поражается при синдроме Дежерина-Русси, отвечает за обработку и фильтрацию информации от болевых, тактильных и проприоцептивных рецепторов. Поэтому при повреждении нейронов поступающая информация искажается: тактильные сигналы воспринимаются как болевые, что является основной причиной развития постинсультной хронической боли и аллодинии.
Вторым компонентом клиники таламического синдрома выступают нарушения ГАМКергической системы нейромедиаторов, которая в норме угнетает передачу болевых сигналов в соматосенсорную кору. Третья составляющая невропатической боли - центральная сенситизизация, возникающая на фоне гипервозбудимости нейронов зрительного бугра, чрезмерного выделения глутамата, устойчивой деполяризации.
Симптомы таламического синдрома
У половины пациентов клинические проявления возникают в первый месяц после инсульта, 37% больных сталкиваются с симптомами в промежутке от 30 дней до 2 лет после нарушения церебрального кровообращения. В 13% случаев между инсультом и развитием таламического синдрома проходит более 2 лет. У таких пациентов изменяется болевая и температурная чувствительность, нарушается восприятие тактильных импульсов.
Основное проявление таламического синдрома - сильные боли на стороне тела, противоположной очагу поражения. Мучительные ощущения описываются как колющие, жгучие, сжимающие и пульсирующие. Некоторые люди сравнивают их с электрическими разрядами. Хронические боли носят постоянный изматывающий характер, нарушают работоспособность и повседневную активность.
Усиление болевого синдрома наблюдается при движениях, воздействии холода или тепла, эмоциональных потрясениях. Однако эти же факторы способны ослаблять боль у части пациентов. В 70% случаев развивается аллодиния - болевые ощущения, которые возникают в ответ на неболевой раздражитель (тепло, прикосновения). Симптоматика сопровождается контралатеральными нарушениями чувствительности и гомонимной гемианопсией.
Для таламического синдрома характерны приступы насильственного неконтролируемого плача или смеха, вегетативно-трофические нарушения. При обширных повреждениях ядер зрительного бугра на противоположной стороне возникает контрактура пальцев кисти - «рука акушера». При этом основные фаланги умеренно согнуты, средние и концевые фаланги полностью разогнуты. Пальцы руки непрерывно двигаются из-за развития хореоатетозного гиперкинеза.
Осложнения
Хотя таламический синдром не представляет серьезной угрозы для пациента, его симптомы существенно снижают качество жизни. Постоянные сильные боли, которые невозможно устранить привычными анальгетиками, вызывают депрессивные расстройства, неврозы, канцерофобию. На фоне особо мучительных и длительных болевых приступов могут возникать суицидальные мысли.
После инсульта присутствуют другие резидуальные последствия, которые усугубляют клиническую картину повреждения таламуса. Нередко встречается частичный или полный паралич отдельных зон тела, дефекты речи, нарушения зрения и слуха. Несмотря на развитие неврологии, полное восстановление трудовой деятельности возможно только у 10% людей, перенесших инсульт. Около 85% пациентов требуют постоянной медико-социальной поддержки.
Основу для постановки диагноза составляет клинико-неврологическое обследование. Учитывают сведения о перенесенном инсульте, его давности и характере реабилитации. На консультации невролога применяются клинические методы оценки нейропатических болей: визуальная аналоговая шкала, опросники DN4 и painDETECT. Как вспомогательные направления диагностики используются:
- КТ/ МРТ головного мозга. Современные методы нейровизуализации показывают наличие и степень повреждения таламических ядер, другие резидуальные проявления инсульта, состояние коры головного мозга и подкорковых ядер.
- ЭЭГ. Программа комплексного обследования включает оценку функциональной активности головного мозга. По результатам электроэнцефалографии определяются очаги повышенной судорожной готовности, которые типичны для инсультов, структурных и метаболических энцефалопатий.
- Консультация психиатра. Если стандартные обследования не выявляют связи между органическим поражением таламуса и болевым синдромом, необходимо исключить психогенные варианты боли. Диагностика включает клиническую беседу с врачом, тестирование и заполнение опросников.
Признаки таламического синдрома необходимо отличать от других вариантов невропатической боли. Дифференциальную диагностику проводят с рассеянным склерозом, компрессионными невропатиями, постгерпетической невралгией. В ходе расширенного обследования исключают диабетическую нейропатию, ВИЧ-обусловленные невралгии, боли, вызванные травмой или дегенеративными заболеваниями спинного мозга.
Лечение таламического синдрома
Консервативная терапия
Для устранения хронической боли необходима патогенетическая терапия, которая воздействует на механизмы появления симптоматики и повышает активность собственной антиноцицептивной системы. Наиболее изучена эффективность трициклических антидепрессантов, которые в средневысоких дозах уменьшают жгучие и мучительные боли, купируют проявления аллодинии. Применение амитриптилина дает стойкий клинический эффект у 66% пациентов.
Эксперты Европейской федерации неврологических сообществ рекомендуют прегабалин в качестве препарата первой линии при таламическом синдроме. Рассматривается применение других видов антиконвульсантов, однако их эффективность при постинсультных болях клинически не доказана. При недостаточном эффекте от психотропных средств показаны опиоидные анальгетики, которые применяют под строгим врачебным контролем для предупреждения зависимости.
В качестве экспериментальной терапии используются антагонисты NMDA-рецепторов, которые влияют на механизмы болевой памяти и предупреждают хронизацию таламических болей. В комплексном лечении применяют когнитивно-поведенческую психотерапию, которая помогает людям справляться с неприятными симптомами, препятствует развитию постинсультной депрессии. В редких случаях прибегают к транскраниальной магнитной стимуляции.
Хирургическое лечение
При отсутствии эффекта от консервативной тактики и сохранении мучительных болей, мешающих полноценной жизни, оценивают показания к таламотомии. Во время операции выполняют стереотаксическую деструкцию таламических ядер, после которой болевой синдром уменьшается. В нейрохирургии такое вмешательство проводят редко и только в сложных случаях, поскольку после операции есть риски необратимых побочных эффектов.
Прогноз и профилактика
Медикаментозные и другие методы лечения таламических болей имеют ограниченную эффективность, поэтому прогноз сомнительный. Профилактика синдрома Дежерина-Русси заключается в предупреждении его причины - нарушений мозгового кровотока. Необходим регулярный контроль артериального давления, своевременная и комбинированная терапия гипертензии, коррекция липидного состава крови диетотерапией и фармакотерапией.
1. Неврология. Национальное руководство. 2-е издание/ под ред. Е.И. Гусева, А.Н. Коновалова, В.И. Скворцова. - 2018.
3. Центральная постинсультная боль (литературный обзор)/ Н.Ю. Хачаянц// Международный журнал прикладных и фундаментальных исследований. - 2015. - №5.
4. Комплексное лечение болей в клинике нервных болезней при синдроме Дежерина-Русси/ С.А. Гуляев, А.А. Овчинникова, А.В. Овчинников// РМЖ. - 2013. - №35.
Гемоглобинопатии
Гемоглобинопатии - это группа тяжелых наследственных заболеваний крови, обусловленных нарушением структуры гемоглобина или снижением синтеза одной и более глобиновых цепей. Клиническая картина крайне разнообразна. Общими симптомами являются гемолитическая анемия, увеличение селезенки, поражение костей. Диагностика осуществляется с помощью микроскопии мазка периферической крови, электрофореза гемоглобина, генетических исследований. Для лечения применяют переливание компонентов крови, препараты гидроксимочевины, инфузионную терапию. У тяжелых пациентов выполняется спленэктомия, аллотрасплантация стволовых клеток.


Гемоглобинопатии - ряд врожденных гемолитических анемий, характеризующихся изменением аминокислотной последовательности гемоглобина или подавлением образования цепей глобина. Данные патологии нередко заканчиваются летально уже в раннем детском возрасте. Известно около 50 видов гемоглобинопатий. Самыми частыми и опасными для жизни считаются серповидно-клеточная анемия (СКА), талассемии. Гемоглобинопатии распространены на территории Центральной Африки, Южной Азии и наблюдаются преимущественно у лиц негроидной расы. Талассемии также встречаются в странах Средиземноморья. Ежегодно рождается около 350000 детей с дефектами гемоглобина.

Причины гемоглобинопатий
Гемоглобинопатии относятся к аутосомно-рецессивным генетическим заболеваниям. Качественные гемоглобинопатии развиваются вследствие мутаций генов, ответственных за синтез определенных аминокислот в бета-цепи глобина. В результате происходит замена одной аминокислоты на другую (глутаминовой кислоты на валин, лизин и пр.). Это приводит к образованию аномального гемоглобина, гораздо менее растворимого, чем нормальный гемоглобин А, придающего красным кровяным тельцам иную форму (мишеневидную, серповидную), что нарушает их функции и уменьшает продолжительность жизни.
Количественные гемоглобинопатии обусловлены мутацией генов, которые кодируют целую цепь глобина (чаще альфа и бета). При этом сдвигается баланс между глобиновыми цепями - при недостаточном синтезе альфа-цепей возникает избыток бета-цепей, и наоборот. Уменьшаются размеры эритроцитов, в них снижается содержание гемоглобина, а мембрана становится более подверженной различным повреждениям.
Существуют факторы, провоцирующие тяжелые приступы (кризы). К ним относятся обезвоживание, переохлаждения, инфекции, сопровождающиеся высокой лихорадкой. У женщин обострения нередко развиваются на фоне беременности. Но главный патологический стимул качественных гемоглобинопатий - уменьшение концентрации в крови кислорода (гипоксия). Это может произойти, например, при подъеме на большую высоту (восхождение на гору, полет на самолете), где снижено парциальное давление кислорода воздуха, или при тяжелых болезнях дыхательной системы (пневмонии).
Гемоглобинопатии имеют сходные патогенетические механизмы. Измененная структура гемоглобина предрасполагает к интенсивному гемолизу. Длительно текущая анемия способствует компенсаторной гиперплазии костного мозга. Возникает деформация костей черепа, искривление позвоночника. Развиваются экстрамедуллярные очаги кроветворения, приводящие к увеличению размеров печени и селезенки (гепатоспленомегалии). Вследствие спленомегалии наступает гиперспленизм - усиленная деструкция красных клеток крови синусоидами селезенки.
Из-за регулярного гемолиза эритроцитов печень секретирует в желчь большое количество билирубина, что создает условия для образования камней желчного пузыря. У больных гемоглобинопатиями часто возникает перегрузка железом, как за счет постоянных переливаний крови, так и из-за повышения абсорбции железа желудочно-кишечным трактом. Большое количество железа в тканях усиливает процессы перекисного окисления липидов, что повреждает различные органы.
При качественных гемоглобинопатиях под влиянием сниженного содержания кислорода в крови молекулы нерастворимого аномального гемоглобина растягивают мембрану эритроцитов, что приводит к изменению их формы. Деформированные эритроциты хуже переносят кислород, а также способны приклеиваться к сосудистому эндотелию, тем самым закупоривая мелкие сосуды, вызывая тромбозы, окклюзии и инфаркты.
Классификация
Гемоглобинопатии подразделяются на качественные, обусловленные нарушением структуры (последовательности аминокислот) гемоглобина, и количественные, характеризующиеся снижением образования глобиновых цепей. Качественные гемоглобинопатии представлены следующими формами:
- Серповидно-клеточная анемия (Гемоглобинопатия S). Наиболее частый вид. Подразделяется на гомозиготную форму (собственно СКА) с яркой клинической симптоматикой и гетерозиготное носительство (серповидно-клеточную аномалию), имеющее бессимптомное или легкое течение.
- Гемоглобинопатия С. Клиника схожа с СКА, но менее выражена. Отличается большей степенью спленомегалии, чем при СКА.
- ГемоглобинопатияCS (Африканский ревматизм). По течению напоминает СКА. Преобладают приступообразные костно-суставные боли.
- Гемоглобинопатии Е иD. Протекают с небольшой гемолитической анемией.
- Наследственная метгемоглобинемия. При этой разновидности под влиянием различных факторов образуется окисленная форма гемоглобина - метгемоглобин, который более стойко связывается с кислородом и не отдает его тканям.
- Анемия, вызванная носительством нестабильных гемоглобинов. Это доброкачественная патология, при которой возникает незначительная гемолитическая анемия после приема сульфаниламидных препаратов.
К количественным аномалиям гемоглобина относят:
- Бета-талассемии. Наиболее распространенный вариант. Подразделяется на малую талассемию (гетерозиготное носительство с бессимптомным течением или легкой гемолитической анемией) и большую талассемию (анемию Кули) с развернутой тяжелой клинической картиной.
- Альфа-талассемии. Течение может быть различным в зависимости от количества мутантных генов. В основном сходны с гетерозиготной бета-талассемией.
- Синдром водянки плода c гемоглобином Барт (Hb Bart’s). Наиболее тяжелый вид альфа-талассемии. Ребенок погибает внутриутробно.
- ГемоглобинопатиюH. Благоприятная малосимптомная форма альфа-талассемии.
- Бета-дельта-талассемии. Практически неотличима от бета-талассемии.
- ГемоглобинопатиюLepore. Эта форма развивается вследствие слияния бета-цепей глобина и схожа с бета-талассемией.
Симптомы гемоглобинопатий
Клиническая картина гемоглобинопатий разнообразна. Гетерозиготные больные имеют либо бессимптомное, либо легкое течение. Гомозиготные формы начинают себя проявлять уже с раннего детства (6 месяцев - 1 год). Общая симптоматика включает признаки гемолитической анемии (бледность, желтушность кожи и слизистых, увеличение селезенки), патологию развития скелета - башенный череп (четырехугольный), уплощенная переносица, искривленный позвоночник.
Из-за повышенной секреции билирубина в желчь могут беспокоить симптомы желчнокаменной болезни уже в детском возрасте - тяжесть или ноющие боли в правом подреберье, приступы желчной колики, обесцвечивание кала. На коже голеней часто возникают длительно незаживающие язвы. Для болезней с дефектом в структуре гемоглобина характерно кризовое течение. Самые тяжелые приступы, нередко заканчивающиеся летально, встречаются при серповидно-клеточной анемии.
Вазоокклюзивный криз
Наиболее типичный. Происходит закупорка мелких сосудов различных органов. Дети испытывают боли в длинных трубчатых костях, у них отекают кисти, стопы, что затрудняет движения лучезапястных, голеностопных суставов (hand-foot синдром). Микротромбозы сосудов кишечника вызывают абдоминальные боли. У молодых мужчин нередко развивается приапизм, впоследствии приводящий к эректильной дисфункции. Кризу сопутствует лихорадка, тахикардия, потливость.
Гемолитический криз
При гемолитическом кризе происходит массивное разрушение красных телец с резким снижением содержания гемоглобина, эритроцитов в крови. Усиливается имеющаяся желтушность, бледность кожных покровов, слизистых, возникают лихорадка, поясничные и абдоминальные боли, присоединяются симптомы снижения артериального давления (головокружение, обмороки). Моча приобретает темный цвет за счет большого количества гемоглобина.
Секвестрационный криз
Во время секвестрационного криза также стремительно падает уровень гемоглобина. Но это происходит не из-за гемолиза, а вследствие венозного тромбоза и скопления большого количества крови в расширенных синусоидах печени и селезенке, что и приводит к обеднению общего кровотока. Кроме симптомов тяжелой анемии по причине выраженной гепатоспленомегалии появляются сильные и распирающие боли в левом и правом подреберье.
Апластический криз
Достаточно редкая форма криза. Она развивается при инфицировании парвовирусом В19, который способен угнетать костномозговое кроветворение. Это сопровождается стремительным (но обратимым) снижением концентрации не только эритроцитов, но и всех других клеток периферической крови (лейкоцитов, тромбоцитов). Поэтому к признакам анемии присоединяется геморрагический синдром (кровотечения из носа, десен), различные инфекции (в основном ОРВИ).
Общими осложнениями гемоглобинопатий считаются ЖКБ, патологические переломы длинных трубчатых костей. Гетерозиготные формы редко сопровождаются неблагоприятными событиями, так как имеют легкое течение. При количественных гемоглобинопатиях из-за отложения избыточного количества железа во внутренних органах развивается сердечная недостаточность, цирроз печени, сахарный диабет 2 типа.
Качественные патологии гемоглобина характеризуются широким спектром неблагоприятных последствий. Наиболее опасными считаются эмболия легочных сосудов, инфаркт миокарда, ОНМК, которые примерно у 10% пациентов приводят к смерти. Закупорка микрососудов, питающих кости, ведет к асептическому некрозу головки бедренной кости (АНГБК). Вследствие постоянных инфарктов селезенки возникает функциональный асплезнизм, из-за чего часто развиваются бактериальные инфекции (бронхиты, пневмонии) с тяжёлым течением, нередко с летальным исходом.
Курацией больных с гемоглобинопатиями занимаются врачи-гематологи, генетики. Во время общего осмотра обращается внимание на цвет кожных покровов (бледность, желтуха), конституциональные нарушения (задержку нервно-психического, физического развития ребенка, аномалии строения скелета). Дополнительное обследование включает:
Гемоглобинопатии дифференцируют с другими врожденными гемолитическими анемиями (мембранопатиями, ферментопатиями, микросфероцитарной анемией Минковского-Шоффара). Постоянные тромбозы нужно дифференцировать от различных тромбофилий. Перегрузку железом следует отличать от наследственного гемохроматоза. Анемия, оссалгии требуют исключения злокачественных миелопролиферативных заболеваний.
Лечение гемоглобинопатий
Людям, страдающим гемоглобинопатиями, требуется проведение сложной многокомпонентной терапии, поэтому все пациенты с гомозиготными формами подлежат обязательной госпитализации в гематологический стационар. Больным с гетерозиготными формами лечение не показано. Основные принципы ведения качественных и количественных патологий гемоглобина несколько отличаются друг от друга.
Подбор консервативной терапии производится с учетом вида гемоглобинопатии, течения заболевания, наличия тех или иных осложнений. Оценивается как клиническая симптоматика, так и лабораторные данные (главным образом, показатели красной крови). Выделяют следующие направления лечения:
- Купирование кризов. При вазоокклюзивных кризах используются обезболивающие препараты (нестероидные противовоспалительные средства), а при выраженном болевом синдроме - наркотические анальгетики. Также для подавления преципитации деформированных эритроцитов назначаются ингаляции кислородом, пероральная или внутривенная регидратация.
- Предупреждение кризов. Для постоянного приема больным качественными гемоглобинопатиями назначается гидроксимочевина. Этот препарат стимулирует образование фетального гемоглобина, подавляющего экспрессию гена, ответственного за синтез аномальных нерастворимых гемоглобинов, что уменьшает склонность эритроцитов к деформации, снижает частоту кризов.
- Терапия осложнений. Инфекционные осложнения до получения результатов бактериологических исследований лечат антибактериальными препаратами, активными против пневмококка, гемофильной палочки, менингококка. Используются антибиотики из группы пенициллинов (амоксициллин). При развитии тромбозов применяются антикоагулянты (низкомолекулярные, нефракционированные гепарины).
- Гемотрансфузии. Так как анемия у пациентов с количественными патологиями гемоглобина всегда тяжелая, основу лечения составляют регулярные гемотрансфузии. Людям, страдающим качественными гемоглобинопатиями, переливания крови проводятся только при секвестрационных, гемолитических, а также апластических кризах.
- Борьба с перегрузкой железа и дефицитом фолатов. Для выведения избытка железа из организма используются хелатирующие средства (дефероксамин). Этот препарат обычно назначается вместе с аскорбиновой кислотой, так как она потенцирует хелатирующее действие дефероксамина. Из-за постоянного гемолиза у больных повышен расход фолатов, поэтому им показан длительный прием больших доз фолиевой кислоты.
Для ряда пациентов с выраженными признаками гемолиза эффективным лечением является спленэктомия - оперативное удаление селезенки. Другой хирургический вид лечения, позволяющий добиться полной ремиссии - аллотрансплантация гемопоэтических стволовых клеток. Однако этот метод применяется редко, только в очень тяжелых случаях, так как он сопряжен с высокой частотой летальных исходов. При холелитиазе выполняется холецистэктомия.
Экспериментальное лечение
В настоящее время ведутся клинические исследования по поиску лекарства для излечения гемоглобинопатий. Есть успешные результаты генной терапии СКА. Суть лечения заключается во введении в стволовые клетки пациента гена, кодирующего синтез нормальной бета-глобиновой цепи, с помощью обезвреженного лентивируса. Препарат называется LentiGlobin BB305. Его применение привело к улучшению показателей крови, что позволило отказаться от постоянной стандартной терапии. Также проводятся испытания данного препарата при бета-талассемии.
Гемоглобинопатии являются тяжелыми заболеваниями с опасными жизнеугрожающими осложнениями. Пациенты с гомозиготной альфа-талассемией умирают еще до рождения в утробе матери. Больные бета-талассемией погибают до наступления пубертатного возраста от сердечной недостаточности. Люди, имеющие качественные гемоглобинопатии, при грамотном лечении могут прожить дольше 50 лет. Основная причина смерти - бактериальные инфекции, тромботические осложнения. При гетерозиготных формах заболевания в большинстве случаев продолжительность жизни не отличается от общей популяции.
Первичная профилактика проводится среди семей, имеющих высокий риск развития гемоглобинопатий. Она заключается в пренатальной диагностике с прерыванием беременности по медпоказаниям. Пациенты, страдающие качественными аномалиями гемоглобина, обязательно должны получить вакцину от гемофильной палочки, пневмококка, менингококка. Детям от 4 месяцев до 6 лет показано длительное применение пенициллиновых антибиотиков для профилактики инфекций. То же касается больных, перенесших спленэктомию.
Читайте также:
- Кардиогенный шок. Классификация кардиогенного шока ( Чазова ). Клиника кадиогенного шока.
- Вред органической (угольной) пыли. Пыль как причина пневмоний и пневмомикозов
- Цели и задачи программы подготовки нейрохирургов в ординатуре
- Анализы для исследования иммунитета. Что проверять?
- Минералокортикоидные гормоны и гормоны щитовидной железы при заболеваниях легких.