Лучевые признаки поражения легких при синдроме Берта-Хога-Дьюба
Добавил пользователь Skiper Обновлено: 22.01.2026
Дифференциальная диагностика:
Рак почки другой этиологии, а также синдром Марфана, синдром Элерса-Данло, наследственный дефицит альфа1-ингибитора протеаз, кисты в легких.
Результат исследования:
• Мутация не выявлена.
• Мутация выявлена в гетерозиготном состоянии.
• Мутация выявлена в гомозиготном состоянии.
• Мутация выявлена в компаунд -гетерозиготном состоянии.
Описание
Метод определения Секвенирование. Выдаётся заключение врача-генетика.
Исследуемый материал Цельная кровь (с ЭДТА).
Исследование мутаций в гене FLCN.
Тип наследования.
Аутосомно-доминантный.
Гены, ответственные за развитие заболевания.
FLCN - ген фолликулина (FOLLICULIN). Ген расположен на хромосоме 17 в регионе 17p11.2. Содержит 14 экзонов, кодирующими являются экзоны с 4-го по 14-й.
Мутации в данном гене приводят также к развитию первичного спонтанного пневмоторакса. Первичный спонтанный пневмоторакс может быть частью клинического спектра синдрома Бёрта-Хоба-Дьюба, при неполной пенетрантности гена.
Мутации в данном гене приводят также к развитию соматического колоректального рака и соматической хроматофобной почечной карциноме.
Определение заболевания.
Синдром относится к редко встречающимся заболеваниям, и ассоциирован с развитием фиброфолликулом (доброкачественные опухоли волосяного фолликула), кистами в легких и повышенным риском возникновения рака почки и толстого кишечника. У пациентов с синдромом BHD риск развития рака почки составляет 15-30%.
Патогенез и клиническая картина.
Возникают доброкачественные опухоли волосяного фолликула, кисты в легких, часто - возникновения рака почки и толстого кишечника.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Список литературы
• Birt, A. R., Hogg, G. R., Dube, W. J. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Derm. 113: 1674-1677, 1977.
• Happle, R. Hornstein-Birt-Hogg-Dube syndrome: a renaming and reconsideration. Am. J. Med. Genet. 158A: 1247-1251, 2012.
• OMIM.
Подготовка
Специальной подготовки к исследованию не требуется. Обязательны к заполнению:
• анкета генетического исследования *.
•.
• информированное согласие.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Показания к применению
Типичная клиническая картина.
Кого надо обследовать при выявленной мутации:
При выявлении у ребенка - обоих родителей, братьев и сестер.
Источник: Invitro.
Бёрта-Хога-Дьюба синдром
Синдром Birt-Hogg-Dube относится к редко встречающимся заболеваниям, и ассоциирован с развитием фиброфолликулом (доброкачественные опухоли волосяного фолликула), кистами в легких и повышенным риском возникновения рака почки и толстого кишечника. У пациентов с синдромом BHD риск развития рака почки составляет 15-30%.
В Центре Молекулярной Генетики разработана методика поиска мутаций в гене фолликулина FLCN (OMIM 607273), ответственных за развитие аллельной серии заболеваний: синдрома Birt-Hogg-Dube (BHD) (OMIM 135150) и первичного спонтанного пневмоторакса (PSP) (OMIM 173600). Ген FLCN локализован в локусе 17p11.2. и состоит из 14 экзонов, но кодирующими являются экзоны с 4-го по 14-й. Заболевания наследуются по аутосомно-доминантному типу. В экзоне 11 гена FLCN исследователями описан участок poly-С(8) с высокой мутационной активностью. Возникающие в этом районе делеции (с.1285delC) и инсерции (с.1285insC) приводят к сдвигу рамки считывания и возникновению преждевременного стоп-кодона, что влечет за собой выработку неполноценного, укороченного белка.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Синдром Бёрта-Хога-Дьюба, ген FLCN м.
FLCN - ген фолликулина (FOLLICULIN). Ген расположен на хромосоме 17 в регионе 17p11.2. Содержит 14 экзонов, кодирующими являются экзоны с 4-го по 14-й.
Мутации в данном гене приводят также к развитию первичного спонтанного пневмоторакса. Первичный спонтанный пневмоторакс может быть частью клинического спектра синдрома Бёрта-Хоба-Дьюба, при неполной пенетрантности гена.
Мутации в данном гене приводят также к развитию соматического колоректального рака и соматической хроматофобной почечной карциноме.
Синдром относится к редко встречающимся заболеваниям, и ассоциирован с развитием фиброфолликулом (доброкачественные опухоли волосяного фолликула), кистами в легких и повышенным риском возникновения рака почки и толстого кишечника. У пациентов с синдромом BHD риск развития рака почки составляет 15-30%.
Возникают доброкачественные опухоли волосяного фолликула, кисты в легких, часто - возникновения рака почки и толстого кишечника.
Синдром Жильбера - симптомы и лечение
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Жильбера — это генетический пигментный гепатоз с аутосомно-доминантным типом наследования, протекающий с повышением уровня неконъюгированного (свободного) билирубина, чаще проявляющееся в период полового созревания и характеризующийся доброкачественным течением [1] .
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
К провоцирующим факторам проявления синдрома можно отнести:
- голодание или переедание;
- жирную пищу;
- некоторые лекарственные средства;
- алкоголь;
- инфекции (грипп, ОРЗ, вирусный гепатит);
- физические и психические перегрузки;
- травмы и оперативные вмешательства.
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
- желтуха различной степени выраженности;
- ксантелазмы век (жёлтые папулы);
- периодичность появления симптомов [1] .
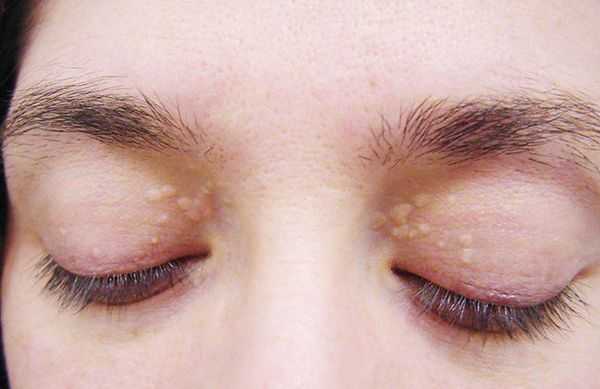
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
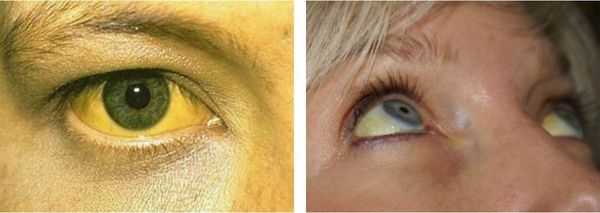
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
- слабость;
- недомогание;
- подавленность;
- плохой сон;
- снижение концентрации внимания.
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
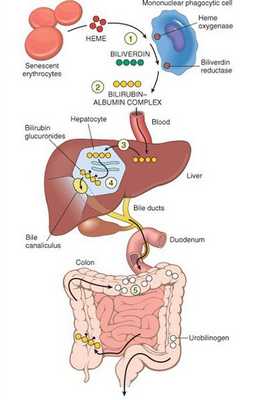
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами ("жирами") клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
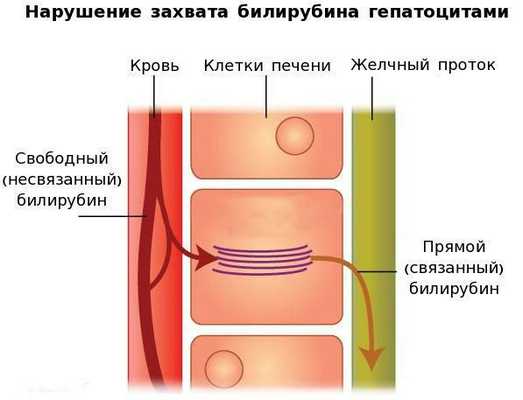
В конечном итоге вышеперечисленные патологические процессы приводят к увеличению содержания свободного (несвязанного) билирубина в плазме, что обуславливает клинические проявления заболевания [6] .
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Читайте также: