Мукополисахариды в слизистой рта. Содержание мукополисахаридов при потере зубов
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Ферментная заместительная терапия - лечение, заключающееся в пожизненном введении препарата (рекомбинантного энзима) пациентам с врожденным дефектом метаболизма.
1. Краткая информация
1.1 Определение
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Мукополисахаридоз VI типа (Синдром Марото Лами) Наследственная лизосомальная болезнь накопления, при которой недостаточность фермента N-ацетилгалактозамин-4-сульфатазы (арилсульфатазы В) приводит к нарушению ступенчатой деградации глюкозаминогликана (ГАГ) дерматансульфата 1.
Синдром Марото-Лами является клинически неоднородным заболеванием с точки зрения распространенности и скорости прогрессирования поражения различных органов и систем . Болезнь характеризуется отставанием в росте, изменениями со стороны органов зрения, огрубением черт лица, снижением слуха, тугоподвижностью в суставах, гепатоспленомегалией, постепенным развитием сердечно-сосудистой и дыхательной недостаточности. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу.
1.2 Этиология и патогенез
Недостаточность фермента N-ацетилгалактозамин-4-cульфатазы (арилсульфатазы В) приводит к нарушению ступенчатой деградации гликозаминогликана (ГАГ) дерматансульфата. ГАГ накапливается внутри лизосом и обусловливает клиническую картину тяжелого хронического прогрессирующего заболевания. Недостаточность арилсульфатазы В обнаруживается во всех тканях, в том числе в культуре фибробластов. Ген ARSB, кодирующий арилсульфатазу В, локализуется в хромосомной области 5q14.
Синдром Марото — Лами наследуется по аутосомно-рецессивному типу [3].
1.3 Эпидемиология
МПС VI типа встречается с популяционной частотой 1:300 000.
1.4 Кодирование по МКБ-10
E 76.2 - Мукополисахаридоз VI типа
1.5 Примеры диагнозов
Мукополисахаридоз VI типа (синдром Марото-Лами). Органическое поражение головного мозга, внутренняя гидроцефалия в стадии компенсации, состояние после наложения вентрикулоперитонеального шунта от 2007г. Слепота вследствие тотального помутнения роговицы обоих глаз и частичной атрофии зрительных нервов. Расходящееся косоглазие. Диспраксия походки и мелкой моторики рук. Сужение позвоночного канала в шейном отделе, компрессия спинного мозга в шейном отделе. Вторичная кардиомиопатия. Недостаточность митрального и аортального клапана. НК 2А степени. Синдром обструктивного апноэ сна тяжелой степени. Деформация позвоночника. Деформация грудной клетки. Сгибательные контрактуры локтевых и коленных суставов. Деформация лучезапястных суставов. Плоскостопие комбинированное 2 ст. Аденоиды 2-3 степени. Гипоплазия зубной эмали. Пупочная грыжа.
1.6 Классификация
В соответствии с дефицитом / отсутствием метаболических лизосомальных ферментам и соответствующим генным дефектам и тяжести клинической симптоматики выделяют несколько типов мукополисахаридозов (табл.1).
Таблица 1 - Классификация (номенклатура) МПС.
Дефицит или отсутствие идуронат-2-сульфатазы
Дефицит или отсутствие сульфоидуронат сульфатазы
Дефицит гепаран-?-глюкозаминид N-ацетилтрансферазы
Дефицит N-ацетилглюкозамин-6- сульфатазы
В настоящее время выделяют 3 формы Мукополисахаридоза VI типа (синдрома Марото-Лами): при тяжелой форме дебют заболевания происходит в возрасте 1-3 года, при среднетяжелой проявления начинают беспокоить с 6 лет, при легкой - после 20 лет.
1.7 Клиническая картина
Синдром Марото-Лами является клинически неоднородным заболеванием с точки зрения распространенности и скорости прогрессирования поражения различных органов и систем (Приложение Г1). Болезнь характеризуется отставанием в росте, изменениями со стороны органов зрения, огрубением черт лица, снижением слуха, тугоподвижностью в суставах, гепатоспленомегалией, постепенным развитием сердечно-сосудистой и дыхательной недостаточности 2.
Основными признаками болезни являются грубые черты лица при сохраненном интеллекте, контрактуры суставов различной степени выраженности, поражение сердечно-сосудистой системы, помутнение роговицы.
Внешний вид пациента характеризуется отставанием в росте (90-100 см при быстром прогрессировании; максимальный - 150 см), диспропорциональным телосложением - карликовостью с укорочением туловища. Отмечается изменение формы лица - большой нос с запавшей переносицей, пухлые губы, маленькие зубы с широкими зубными промежутками, позднее прорезывание зубов, макроглоссия. Возможна глухота, пупочная грыжа, паховая грыжа, уплотнение и утолщение кожи, грубые волосы, умеренный гирсутизм. На поздних стадиях развивается глухота, слепота.
Костная система: наблюдаются множественные дизостозы, умеренная тугоподвижность практически во всех суставах, сгибательные контрактуры межфаланговых суставов и клешневидная деформация кисти. Изменения тазобедренных суставов (дисплазия головки бедренной кости), деформация эпифизов бедренных костей с двух сторон ведет к прогрессирующей инвалидизации. Килевидная грудная клетка с широкими ребрами, дефект развития тел позвонков с передним переломом; Х-образное искривление ног; при рентгенографии - точечные пястные кости.
Органы дыхания: частые респираторные инфекции (риниты, отиты). Гипертрофия миндалин и аденоидов, увеличение языка, утолщение надгортанника и голосовых связок, обусловливают развитие дыхательных нарушений разной степени тяжести, включая обструктивное апноэ сна. Особенности деформаций грудной клетки (жесткая грудная клетка в сочетании с кифосколиозом и поясничным лордозом) способствует развитию рестриктивных дыхательных нарушений.
Органы зрения: отмечается помутнение роговицы, связанное с ее утолщением и увеличением (мегалокорнеа), ретинопатия, изменения диска зрительного нерва, внутриглазная гипертензия, глаукома. Поражение зрительного нерва может быть обусловлено отложением гликозаминогликанов в ганглиозных клетках зрительного нерва, компрессией зрительного нерва утолщенной твердой мозговой оболочкой или сужением костных структур вдоль тракта зрительного нерва, а также повышенным внутричерепных давлением. В случае изменения зрительного нерва или повышения внутриглазного давления следует провести исследование полей зрения. Зрительные вызванные потенциалы могут исследоваться для определения функции и сохранности зрительных нервов при затруднении фундоскопического исследования зрительного нерва выраженным помутнением роговицы.
Центральная нервная система: интеллект и поведенческие реакции у больных с данной нозологией обычно не страдают, однако высок риск развития миелопатии шейного отдела позвоночника. Возможно развитие компрессии спинного мозга вследствие сопутствующих нарушений опорно-двигательного аппарата. Описаны случаи сдавления спинного мозга, вызванного утолщением его оболочек или нестабильностью атлантоаксиального сустава (сопровождаются нарушением походки, мышечной слабостью, неуклюжестью при сохранных моторных навыках и дисфункцией мочевого пузыря).
Тяжелая форма болезни сопровождается открытой (сообщающейся) гидроцефалией, нарушениями резорбции спинномозговой жидкости (СМЖ), которые также вносят вклад в развитие неврологических нарушений
Признаки гидроцефалии зачастую появляются медленно и незаметно, и могут заключаться в изменении поведения, возникновении головной боли, нарушений зрения.
У пациентов с тяжёлой формой заболевания иногда наблюдаются судороги. Они могут иметь как фокальный, так и генерализованный характер. Появление судорог требует проведения оценки неврологического статуса. При прогрессировании заболевания часто наблюдаются генерализованные тонико-клонические пароксизмы.
Карпальный тоннельный синдром - нейропатия сдавления у пациентов с различными видами МПС. При отсутствии лечения может привести к необратимой контрактуре дистальных межфаланговых суставов, а также к нарушению или потере чувствительности первых 3 пальцев и к парезу мышц тенара.
Костные изменения при МПС VI типа приводят к снижению подвижности нижней челюсти, что ограничивает способность открывать рот и жевать. Нарушения глотания встречаются редко, отмечаются при среднетяжелом и тяжелом течении заболевания и, в основном, связаны с нарушением функционирования стволовых отделов головного мозга.
У пациентов наблюдаются признаки псевдобульбарного или бульбарного параличей. Симптомы дебютируют с редких поперхиваний твердой пищей, развиваются постепенно и в конечном итоге приводят к грубому нарушению функции глотания или полному ее исчезновению. Отсутствие правильной регуляции акта глотания приводит к забросу пищи, слюны в трахею и бронхиальное дерево и развитию вторичной инфекции в виде рецидивирующих аспирационных бронхитов и пневмоний. Это усугубляет дыхательные нарушения, являющиеся следствием отложения мукополисахаридов в верхних и нижних дыхательных путях.
Сердечно - сосудистая система: отмечается прогрессирующая дегенерация клапанов сердца с образованием стеноза (наиболее часто митрального и аортального). Проявления сердечно-сосудистых нарушений отмечаются с раннего возраста. Большинство пациентов имеют, по крайней мере, один признак ко второму десятилетию жизни. Кардиомиопатия отмечается нечасто.
Желудочно-кишечная система: синдром раздраженного кишечника, гепатоспленомегалия.
2. Диагностика
Диагноз МПС VI устанавливается на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа 5.
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб следует обратить внимание на следующие жалобы и анамнестические события:
Мукозит - симптомы и лечение
Что такое мукозит? Причины возникновения, диагностику и методы лечения разберем в статье доктора Абдуллаевой Айтан Измировны, детского стоматолога со стажем в 8 лет.
Над статьей доктора Абдуллаевой Айтан Измировны работали литературный редактор Юлия Липовская , научный редактор Пётр Козлов и шеф-редактор Маргарита Тихонова

Определение болезни. Причины заболевания
Мукозит (mucositis) — это поражение слизистых оболочек ЖКТ в виде воспаления и язв, которое развивается из-за лучевой или химиотерапии при лечении рака. Появляется чаще в области рта и глотки: в основном поражает слизистую оболочку мягкого нёба, боковой границы языка, стенки глотки, а также слизистую губ, щёк, средней части языка и дна полости рта [1] [2] .

Мукозитом также называют воспаление мягких тканей вокруг зубного импланта, другое его название периимплантный мукозит. Но в этой статье речь пойдёт только об осложнении противоопухолевого лечения.
Это изнуряющее состояние, которое поражает более 40 % пациентов, получающих лучевую или химиотерапию [3] [4] . В тяжёлых случаях мукозит может быть настолько болезненным, что человек не может нормально питаться и принимать лекарства. Это не только ухудшает качество жизни, но и вынуждает врача уменьшить дозу препаратов или облучения или отложить курс, что снижает эффективность противоопухолевого лечения. Кроме того, поражение слизистой оболочки может привести к развитию инфекции.
Причины и факторы риска развития мукозита
Основная причина — ослабление иммунной системы, вызванное противоопухолевым лечением.
При лучевой и химиотерапии погибают не только быстро делящиеся раковые клетки, но и нормальные клетки, которые делятся более активно, например во рту или костном мозге. Поэтому и возникают такие побочные эффекты, как снижение иммунитета, мукозит, усталость, ухудшение аппетита и др.
Препараты, которые используются для лечения рака и могут привести к развитию мукозита:
- Антиметаболические и алкилирующие препараты. Они подавляют синтез ДНК, из-за этого эпителий слизистой оболочки не восстанавливается и, как правило, возникает мукозит.
- Противоопухолевые антибиотики, такие как Адриамицин. Агрессивно влияют на слизистые оболочки и подавляют выработку слюны, что способствует проникновению инфекции в слизистые оболочки у пациентов, ослабленных после лучевой и химиотерапии.
- Противоопухолевые препараты на основе алкалоидов растений (например, Винбластин или Винкристин). Они редко напрямую повреждают слизистую оболочку полости рта, но могут ослабить защитные свойства слизистой за счёт токсинов [3][4][5] .
Чтобы увеличить эффективность терапии, часто увеличивают её интенсивность. Но так как химиотерапия токсична и приводит к серьёзным побочным эффектам, её применение ограничено: приходится уменьшать дозу препарата, что снижает реакцию опухолей на лечение [6] [7] .
При лучевой терапии побочные эффекты наблюдаются только в области облучения. В большинстве случаев пациенты выздоравливают через 2-3 недели после окончания лучевой терапии [1] [5] [11] .
Тяжесть мукозита после лучевой и химиотерапии будет зависеть от множества факторов, включая дозу лекарства, интервал между дозами, тип облучения и объём обрабатываемой ткани.
К факторам риска развития мукозита также можно отнести курение, злоупотребление алкоголем, обезвоживание, заболевания почек, ВИЧ-инфекцию, СПИД, постоянные травмы слизистой из-за протезов, сухость во рту и низкий уровень лейкоцитов в крови [8] [27] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы мукозита
Симптомы мукозита могут возникнуть через 1-2 недели после лучевой или химиотерапии [8] [28] . Первый признак мукозита — эритема (покраснение), затем на месте эритемы появляются эрозии и язвы.

При язве нарушается целостность всех слоёв слизистой оболочки. Так как в слизистой много нервных волокон и окончаний, то её повреждение вызывает сильную боль. Она может быть настолько сильной, что человек не может нормально питаться и разговаривать. Это наиболее тяжёлая форма мукозита, обычно она наблюдается через 5-7 дней после приёма лекарств для химиотерапии [9] [10] .
![Язва в полости рта [25]](https://probolezny.ru/media/bolezny/mukozit/yazva-v-polosti-rta-25_s.jpg)
Язвы обычно покрыты псевдомембраной. Эта мембрана состоит из эпителиальных клеток и разрушенного белка фибрина. Она выглядит как белый, непрозрачный налёт, но если присоединяется инфекция, может казаться желтоватой или зеленоватой. Еда, напитки или местные лекарства также могут окрашивать псевдомембрану в жёлтый или зелёный цвет. При тщательном осмотре часто обнаруживается слегка приподнятая мембрана над уровнем подлежащей слизистой оболочки. Из-за псевдомембран мукозит можно принять за кандидоз.
![Псевдомембрана на язве [26]](https://probolezny.ru/media/bolezny/mukozit/psevdomembrana-na-yazve-26_s.jpg)
Жёсткая пища может травмировать псевдомембрану, и если мембрана отделяется от слизистой, возникает кровотечение.
При облучении и химиотерапии эпителий разрушается и не успевает восстанавливаться, из-за этого слизистая оболочка истончается [11] [12] .
При поражении слизистой кишечника или мочеполовых путей может беспокоить диарея и боль при мочеиспускании.
Патогенез мукозита
У мукозита можно выделить четыре фазы развития:
- Сосудистая (воспалительная) фаза.
- Эпителиальная фаза.
- Язвенная (бактериологическая) фаза.
- Фаза восстановления [2][4][7][13] .

1. Сосудистая (воспалительная) фаза. В первую фазу лучевая или химиотерапия напрямую повреждают клетки эпителия. В ответ на повреждение вырабатываются провоспалительные цитокины, которые запускают воспалительный процесс. Цитокины усиливают разрушение тканей, расширяют кровеносные сосуды и повышают проницаемость их стенок. На этой фазе никаких симптомов на слизистой оболочке ещё нет.
2. Эпителиальная фаза. Начинается на 4-5 день противоопухолевого лечения. Из-за действия цитокинов замедляется репликация (удвоение) эпителиальных базальных клеток, поэтому снижается скорость обновления эпителия. К концу фазы появляется эритема и чувство жжения.
3. Язвенная (бактериологическая) фаза. Начинается примерно на седьмые сутки. Клетки слизистой отмирают и начинают медленнее обновляться, из-за этого слизистая оболочка рта истончается и разрушаются связи между эпителием и подслизистым слоем. Фаза характеризуется выраженными клиническими проявлениями с образованием язв на слизистой оболочке. Язвы инфицируются из-за травмы эпителия.
4. Фаза восстановления. Если у пациента не снижен уровень лейкоцитов и тромбоцитов в крови и курс противоопухолевого лечения завершён, выздоровление наступает через 2-3 недели после начала терапии . Крупные и глубокие либо инфицированные язвы обычно заживают на 1-2 недели дольше. В зависимости от степени повреждения слизистая оболочка на этой фазе может казаться бледной и истончённой.
Чем медленнее развивается мукозит и чем дольше не заживают язвы на слизистой, тем тяжелее будет течение болезни. Чтобы лучше изучить механизм развития мукозита, необходимы дополнительные данные об эпидемиологии и факторах риска [4] [7] [15] [16] .
Классификация и стадии развития мукозита
В Международной классификации болезней 10-го пересмотра (МКБ-10) мукозит полости рта кодируется как K12.3 Оральный мукозит (язвенный). Он делится на несколько видов:
- БДУ, т. е. неуточнённый;
- медикаментозный;
- лучевой (радиационный);
- вирусный.
Классы мукозита полости рта в соответствии с системой оценок Всемирной организации здравоохранения (ВОЗ):
- 0 — признаков мукозита нет;
- I — безболезненная язва, эритема или лёгкая чувствительность;
- II — болезненная эритема или язва, которые не мешают пациенту принимать пищу;
- III — сливающиеся болезненные язвы, которые мешают пациенту принимать твёрдую пищу;
- IV — тяжёлые симптомы, при которых приходится вводить питание через зонд или внутривенно [1][4][10][16] .
Осложнения мукозита
Психологические проблемы. Из-за онкологического заболевания люди часто не могут работать и заниматься активным хобби, реже встречаются с друзьями и родными. Все их планы перестраиваются под болезнь. Если к основном диагнозу присоединяются осложнения терапии, например мукозит, то психологическое состояние пациента может ещё больше ухудшиться. Человек постепенно теряет силы и самостоятельность [17] [18] .
Перерыв в лечении или изменение режима. Если из-за лучевой или химиотерапии развивается тяжёлая степень мукозита, врачу приходится уменьшить дозу препаратов или радиации или отложить курс, что снижает эффективность лечения.
![Тяжёлая степень мукозита [24]](https://probolezny.ru/media/bolezny/mukozit/tyazhyolaya-stepen-mukozita-24_s.jpg)
Вторичная инфекция. Через поражённую слизистую оболочку полости рта в организм, ослабленный после лучевой или химиотерапии, могут попасть патогенные микроорганизмы. В этом случае на фоне мукозита развивается вторичная бактериальная, грибковая или вирусная инфекция. Проведённые исследования показывают, что у 29,1 % пациентов, проходящих лечение от рака, был выявлен простой герпес, который усугублял язвенный мукозит полости рта [3] [12] .
Сепсис. Если вторичную инфекцию не лечить, патогенные микроорганизмы могут массово попасть в кровоток и распространиться по всему организму. Такая тяжёлая системная инфекция называется сепсисом. Он может стать причиной смерти [17] [18] .

Диагностика мукозита
Диагностика мукозита основана на наличии лучевой или химиотерапии в анамнезе. Также важно, когда появились симптомы мукозита и где в полости рта расположены эрозии и язвы.
Мукозит, вызванный лучевой или химиотерапией, возникает на подвижных слизистых оболочках, т. е. в области щёк, губ и дна полости рта. Он редко поражает тыльную сторону языка, твердое нёбо или дёсны.
Вирусные инфекции, например кандидоз, в отличие от мукозита локальны и затрагивают слизистую оболочку твёрдого нёба, десневой и дорсальный язык. Кроме этого, их начало часто совпадает с лихорадкой. Чтобы отличить мукозит от вирусной инфекции, рекомендуется взять мазок со слизистой рта и определить с помощью бактериального посева или цитологического исследования, есть ли на её поверхности инфекция.
Важно оценить тяжесть мукозита, чтобы определить токсичность проводимого противоопухолевого лечения и дальнейшую тактику ведения пациента. Общепринятой шкалы, которая бы описывала тяжесть мукозита, не существует. Поэтому понять, насколько терапия вредит пациенту, бывает сложно. Чаще всего для описания токсичности терапии рака используют критерии Всемирной организации здравоохранения (ВОЗ) и Национального института рака (NCI). Эти шкалы описывают общее состояние полости рта [5] [6] [19] . Например, согласно шкале ВОЗ, нулевая степень тяжести — это отсутствие симптомов мукозита, а четвёртая соответствует тяжёлым симптомам, при которых пациент не может самостоятельно принимать пищу [20] .
Лечение мукозита
Терапия мукозита включает как местное воздействие (только на слизистую оболочку полости рта), так и системные лекарственные и нефармакологические методы лечения, которые действуют на весь организм.
Цели лечения орального мукозита: обеспечить питание, уменьшить болевой синдром и сухость во рту, предотвратить вторичное инфицирование и оральное кровотечение.
Лечение орального мукозита по большей части направлено на устранение симптомов и облегчение состояния пациента, но также разрабатываются средства, чтобы предотвратить его развитие.
Из симптоматических средств назначают анальгетики, так как мукозит III - IV степени тяжести обычно протекает очень болезненно. Согласно рекомендациям ВОЗ, в зависимости от интенсивности боли должны назначаться как системные, так и местные обезболивающие [19] [21] .
Во многих медицинских центрах онкологическим больным, получающим высокие дозы препаратов для химиотерапии, обычно назначают антибиотики и противогрибковые препараты, чтобы предотвратить вторичные инфекции в период снижения иммунитета.
Прогноз. Профилактика
Мукозит развивается на фоне тяжёлой онкологической патологии, поэтому прогноз зависит от тяжести основного заболевания, состояния пациента, а также от выраженности проявлений самого мукозита.
Чаще всего симптоматическое лечение мукозита помогает улучшить состояние пациента: язвы перестают болеть и распространяться. Как правило, всё заживает к 15-20 дню от начала терапии [1] [22] [23] .
Профилактика мукозита
Риск мукозита ниже, если у пациента здоровые зубы и полость рта. Но как показывает практика, почти у всех пациентов, поступающих для лечения в онкологические и онкогематологические стационары, есть проблемы с зубами или дёснами [2] [3] .
Перед лучевой или химиотерапией рекомендуется:
- вылечить зубы и дёсны, если есть такая необходимость;
- за 10-12 дней до начала противоопухолевой терапии сделать профессиональную гигиену полости рта, в которую входит ультразвуковая чистка, использование Air-flow и полировка пастой.

Во время лучевой или химиотерапии необходимо:
- выполнять рекомендации врача по гигиене полости рта;
- посещать стоматолога для осмотра слизистой оболочки полости рта [28] ;
- не травмировать слизистую оболочку: исключить приём горячей, острой, твёрдой пищи и алкоголя; отказаться от курения; как можно реже пользоваться съёмными зубными протезами; использовать только мягкую зубную щётку [3][6][17] .
Мукозит часто осложняет лечение рака. Стоматологическая подготовка и сопровождение пациента в процессе химиолучевой терапии позволяет не только улучшить качество жизни больных, но и избежать вынужденных перерывов в лечении, повышая тем самым эффективность противоопухолевой терапии [4] [21] .
Мукополисахаридоз
Мукополисахаридозы - это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
Общие сведения
Мукополисахаридозы - группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Позвоночник пациентов с мукополисахаридозом искривлен, из-за чего в положении сидя возникает симптом «кошачьей спины». Отмечается укорочение шеи, высокое расположение лопаток и выстояние нижних ребер. Кисти широкие, напоминающие когтистую лапу. Со временем у больных мукополисахаридозом формируются контрактуры суставов. Вначале поражаются локтевые и плечевые суставы, затем - коленные, тазобедренные и голеностопные. Из-за ограничения движений возникает характерная походка - на цыпочках с полусогнутыми ногами.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Мукополисахаридоз типа II или синдром Хантера чаще выявляется у мальчиков и обычно развивается на 2-3 году жизни. Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и затруднения дыхания из-за деформации лицевого скелета. При этом характерный для мукополисахаридоза типа IH кифоз обычно не выявляется, симптом «кошачьей спины» отрицательный. Пациенты часто страдают от респираторных инфекций (бронхитов, трахеитов, пневмоний). Постепенно развиваются нарушения координации движений, возрастает агрессивность. Настроение неустойчивое, с резкими перепадами.
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина - как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина - без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом - менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагностика
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявления гликозаминогликанов в моче, изучения активности ферментов в клеточных культурах, генетического секвенирования. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный - продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
1. Руководство по педиатрии. Врожденные и наследственные заболевания/ Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р - 2007
Дифференциальная диагностика мукополисахаридозов
О мукополисахаридозах (МПС), редких наследственных заболеваниях обмена веществ из группы лизосомных болезней накопления, в последние годы говорят часто. Еще недавно схожесть симптоматики МПС с другими заболеваниями и недостаточная лабораторная диагностика часто приводили к ошибочным диагнозам и неадекватному лечению. Вместе с тем ранняя диагностика мукополисахаридозов и правильная терапия во многом определяют прогноз заболевания и перспективы пациента.
Основа патогенеза МПС — накопление в органах и тканях патологического субстрата, а именно гликозаминогликанов (ГАГ), или мукополисахаридов, которые являются базовым компонентом соединительной ткани. Избыточное накопление ГАГ приводит к дисфункции клеток, органов и тканей и вызывает разнообразные клинические проявления. МПС относится к мультисистемным заболеваниям, поэтому дифференциальная диагностика данной нозологии актуальна для врачей любых специальностей, особенно педиатрического профиля.

Опытом правильной диагностики МПС (на примере синдрома Хантера как наиболее распространенного типа мукополисахаридоза) поделилась старший научный сотрудник лаборатории редких наследственных болезней у детей Центра редких болезней ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России, кандидат мед. наук Татьяна Подклетнова.
Объективная лабораторная диагностика
Основа диагностики — клинические проявления заболевания. Все типы МПС имеют общие внешние клинические признаки, которые могут и должны вызвать у врача на амбулаторном приеме настороженность в отношении МПС: гаргоилизм, костные изменения, органомегалия и т. д.
Лабораторная диагностика включает три этапа.
1. Количественная и качественная оценка экскреции ГАГ в моче. Неспецифический тест, который с высокой степенью вероятности позволяет заподозрить у пациента МПС, не дает, однако, четкой информации о типе заболевания.
При легких формах заболевания избыточное количество ГАГ в моче может не определяться, следовательно, тест будет ложно-отрицательным. На основании только этого лабораторного анализа ставить окончательный диагноз нельзя!
2. Энзимодиагностика. Измерение активности фермента в лейкоцитах крови. При МПС любого типа снижается уровень определенного фермента в лейкоцитах или плазме крови. В частности, при МПС 2-го типа снижается уровень идуронат-2-сульфатазы в плазме (норма: 250-620 nM/mg/h); МПС 2-го типа: 0-20 nM/mg/h); при МПС 1-го типа — α-L-идуронидазы в периферической крови (норма: 61-176 nM/mg/h; МПС 1-го типа: 0-18 nM/mg/h) и т. д. Однако существует ряд заболеваний, при которых уровень ферментов в крови также может снижаться, например, множественная сульфатазная недостаточность. Необходим третий этап.
3. Молекулярно-генетическая диагностика. Наличие мутаций в том или ином гене дает окончательный ответ и возможность установить объективный диагноз. Так, МПС 1-го типа обусловлен мутацией в гене IDUA, МПС 2-го типа — мутацией в гене IDS и т. д.
Существуют и другие методы диагностики, которые обязательно должны быть предложены семьям, планирующим рождение ребенка, но у которых уже есть случаи МПС среди близких родственников. Речь о пренатальной и предимплантационной диагностике.
Пренатальная диагностика проводится между 10-й и 12-й неделями уже наступившей беременности. Выполняется определение ферментной активности в ворсинах хориона, проводится молекулярно-генетическая диагностика плода. Надежность метода достигает 99 %. У родителей есть возможность при выявлении патологии прервать беременность на малом сроке. Или же принять осознанное решение о рождении ребенка с генетическим заболеванием. В этом случае ранняя пренатальная диагностика дает возможность начать адекватное лечение на самом раннем этапе, что, естественно, влечет за собой совершенно иные, более благоприятные прогнозы относительно качества и продолжительности жизни ребенка.
Предимплантационная диагностика проводится перед искусственным оплодотворением и позволяет избежать рождения ребенка с данной генетической патологией.
Шаг 1. Дифдиагностика с другими видами мукополисахаридозов
В современной классификации существуют 7 основных типов мукополисахаридозов. Наиболее часто (помимо МПС 2-го типа) встречаются МПС 1-го, 3-го, 4-го, 6-го типов.
Особенности МПС 1-го типа (синдром Гурлера, синдром Гурлера — Шейе, синдром Шейе):
- гипертрофия десен, редкие мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки, глаукома;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное носовое дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- задержка психомоторного развития, регресс навыков;
- экскреция гепарансульфата и дерматансульфата с мочой.
МПС 1-го типа имеет аутосомно-рецессивный тип наследования, т. е. заболевание одинаково свойственно лицам мужского и женского пола, в то время как МПС 2-го типа — мужское заболевание, девочки болеют крайне редко. Помутнение роговицы при МПС 2-го типа практически не встречается либо очень «нежное», визуально не видное. При МПС-1 помутнение роговицы видно визуально. Костные деформации и проявления при МПС-1 превалируют, ярче выражены, быстрее прогрессируют.
Особенности МПС 3-го типа (синдром Санфилиппо):
- болеют лица мужского и женского пола;
- ведущим является поражение ЦНС: интеллектуальный дефицит, нарушения поведения и сна, двигательные нарушения, эпилепсия;
- высокий рост, кифосколиоз, деформация грудной клетки, контрактуры крупных и мелких суставов менее выраженные;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция гепарансульфата с мочой.
Наиболее яркое отличие МПС 3-го типа — более выраженная неврологическая симптоматика по сравнению с другими типами МПС, для которых характерен интеллектуальный дефицит.
Например, при МПС-1 и МПС-2 иногда бывает и так, что ребенок изначально развивается в соответствии с возрастом, лишь затем появляются симптомы задержки и происходит регресс. В случае МПС-3 ребенок с раннего детства имеет прогрессирующую задержку развития, нарушения поведения, к которым позже присоединяются другие неврологические проявления: двигательные нарушения, судороги. Что касается соматических проявлений, то и здесь есть отличия: дети с МПС 3-го типа имеют достаточно высокий рост (другим типам свойственна низкорослость); при этом типе мукополисахаридоза не бывает цервикального стеноза и синдрома запястного канала.
Особенности МПС 4-го типа (синдром Моркио):
- изменения черт лица по типу гаргоилизма отсутствуют;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, килевидная деформация грудной клетки, грубые деформации крупных и мелких суставов при отсутствии контрактур;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция кератансульфата с мочой.
Примечательно, что детям с МПС 4-го типа не присущ гаргоилизм. Кроме того, особенностью этого типа МПС являются специфические костные деформации: килевидная деформация грудной клетки, грубая деформация позвоночника, вальгусная деформация конечностей. МПС-4 отличает и абсолютно сохранный интеллект пациента (в отличие от МПС-1 или МПС-2, при которых есть пациенты как с высоким уровнем интеллекта, так и с достаточной грубой задержкой интеллектуального развития).
При ранней постановке диагноза и правильном ведении болезни пациенты с синдромом Моркио могут быть успешно адаптированы в обществе.
Особенности МПС 6-го типа(синдром Марото — Лами):
- гипертрофия десен, редкие, мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция дерматансульфата с мочой.
Этот тип МПС имеет всю симптоматику, характерную для других типов мукополисахаридоза. Из-за видимого помутнения роговицы, грубых костных деформаций, выраженных контрактур суставов МПС 6-го типа похож на тяжелые формы МПС-1 и МПС-4.
Важно отметить, что при данном типе МПС, как и при МПС-4, пациенты имеют сохранный интеллект и при ранней диагностике заболевания, своевременном начале ферментозаместительной терапии могут быть социализированы в обществе.
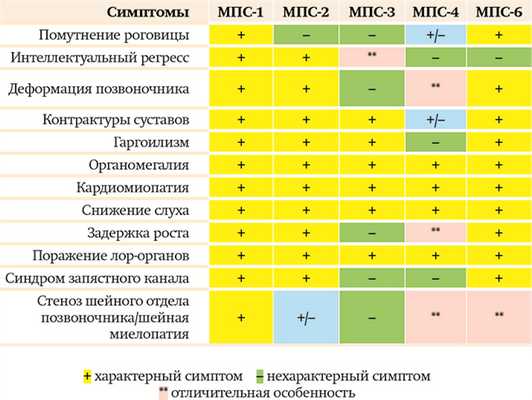
Таблица может помочь врачу на амбулаторном приеме предположить МПС, чтобы потом подтвердить/ опровергнуть диагноз лабораторными исследованиями
Шаг 2. Исключение других болезней накопления
Ряд болезней накопления имеет схожие с МПС внешние клинические признаки. Например, симптоматика альфа-маннозидоза включает:
- грубые черты лица;
- изменения скелета по типу множественного дизостоза;
- поясничный лордоз;
- диффузную мышечную гипотонию;
- шумное носовое дыхание;
- умеренную гепатоспленомегалию;
- нейросенсорную тугоухость;
- пупочную, паховую грыжи;
- умственную отсталость различной степени;
- частые респираторные инфекции, бронхиты, отиты.
Но для альфа-маннозидоза ведущим клиническим признаком является высокая степень нейросенсорной тугоухости, наличие признаков периферической полинейропатии (для МПС-1, МПС-2, МПС-6 характерен только синдром запястного канала), гуморальные и клеточные иммунодефициты. У пациентов с МПС, несмотря на то, что они склонны к частым респираторным инфекциям, иммунодефициты отсутствуют. Но главное отличие в результатах лабораторных исследований. При альфа-маннозидозе отмечается снижение активности альфа-маннозидазы в лейкоцитах. Накопление маннозосодержащих олигосахаридов. ГАГ в моче отсутствует. По результатам ДНК-диагностики заболевание обусловлено мутациями в гене MAN2B1.
Муколипидоз 2-го и 3-го типа также имеет симптомы, которые «роднят» его с разными типами МПС. Муколипидоз 2-го типа имеет более тяжелое течение и клинические проявления практически с первых месяцев жизни, быстрое прогрессирование. Муколипидоз 3-го типа мягче, отличается более поздним проявлением симптоматики и чуть меньшей скоростью прогрессирования. При этом вне зависимости от типа муколипидозу свойственны общие с мукополисахаридозом симптомы:
- грубый гаргоилизм;
- низкорослость;
- помутнение роговицы;
- тугоподвижность суставов;
- интеллектуальный дефицит;
- паховые и пупочные грыжи;
- органомегалия и др.
Отличить одно заболевание от другого помогает лабораторная диагностика. При муколипидозе 2-го типа отмечается повышение уровня лизосомных ферментов (N-ацетилгексозаминидазы) в плазме и низкий уровень в лейкоцитах крови. При муколипидозе 3-го типа — резкое снижение активности N-ацетилглюкозамин-1-фосфотрансферазы в лейкоцитах и культуре кожных фибробластов (ККФ). В отличие от МПС муколипидоз обусловлен мутациями в гене GNPTAB.
Фукозидоз — очень редкая патология, которая имеет общие с МПС симптомы: огрубление черт лица, низкорослость, органомегалию, грыжи, нейросенсорную тугоухость, контрактуры крупных и мелких суставов, помутнение роговицы и т. д.
Как и другие болезни накопления, фукозидоз может протекать с разной скоростью прогрессирования. При тяжелой форме болезни клинические проявления возникают уже на первом году жизни, быстро прогрессируют, приводя к тяжелой инвалидизации и раннему летальному исходу. При мягком течении клинические симптомы появляются в конце первого десятилетия, медленнее прогрессируют.
Главной отличительной особенностью фукозидоза являются специфические кожные проявления — ангиокератома, ангидроз. Наличие ангиокератомы вкупе с характерной для МПС симптоматикой дает основания предполагать фукозидоз. Окончательное слово за лабораторной диагностикой. При фукозидозе отмечается резкое снижение активности α-L-фукозидазы и ККФ, накопление гликолипидов и гликопротеинов в клетках. Молекулярно-генетическая диагностика указывает на мутации в гене FUCA1.
GM1-ганглиозидоз , в отличие от предыдущего заболевания, широко распространен среди болезней накопления. Несмотря на некоторые схожие с МПС симптомы (гаргоилизм, низкорослость, гиперплазия десен, костные деформации, органомегалия, тугоподвижность суставов и т. д.), GM1-ганглиозидоз имеет яркие отличительные особенности. В частности, ему свойственны макулодистрофия по типу «вишневой косточки», поражение почек, быстро прогрессирующее поражение ЦНС, эпилепсия, грубая задержка психомоторного развития, ангиокератома. По результатам лабораторной диагностики отмечается резкое снижение активности β-галактозидазы в лейкоцитах и ККФ. Заболевание обусловлено мутациями в гене GLB1. Болезнь практически всегда имеет тяжелое течение и неблагоприятный исход в первые годы жизни.
Множественная сульфатазная недостаточность — заболевание, которое легко спутать с МПС 2-го типа, если не выполнить объективную лабораторную диагностику. Клиническая симптоматика заболевания почти идентична симптоматике МПС-2, но результаты лабораторных исследований показывают принципиальные отличия. Так, при множественной сульфатазной недостаточности в крови снижен уровень не только идуронат-2-сульфатазы, но и других ферментов, в частности арилсульфатаз А, В, С. ДНК-диагностика показывает мутации в гене SUMF1.
Шаг 3. Исключение иных наследственных заболеваний или синдромальных состояний, схожих с болезнями накопления, в том числе МПС
Микроделеция Xq28, включающая ген IDS. Заболевание обусловлено делецией в хромосомной области, которая затрагивает ген IDS. Мутации этого гена лежат в основе МПС 2-го типа, поэтому неудивительно, что клинические признаки микроделеции Xq28 практически совпадают с симптомами МПС-2, включая высокий уровень ГАГ в моче. Примечательно, что ферментозаместительная терапия, назначаемая обычно при МПС-2, в этом случае также может давать положительную динамику. Но формально это все-таки не МПС-2, поскольку заболевание вызвано делецией, затрагивающей ген IDS, а не мутацией в нем.
Синдром Беквита — Видемана. Достаточно распространенный синдром, имеющий схожую с МПС симптоматику: интеллектуальный дефицит, органомегалию, грыжи. В то же время молекулярно-генетическое исследование показывает совершенно отличную от МПС картину: это синдромальное состояние, обусловленное изменениями в регионе 11р15, включающем гены факторов роста и гены-супрессоры опухолевого роста.
Х-сцепленная демиелинизирующая лейкодистрофия со спондилоэпифизарной дисплазией. Для этого заболевания характерны клинические признаки, схожие с симптоматикой болезней накопления: гаргоилизм, изменения скелета по типу множественного дизостоза (сколиоз, воронкообразная деформация грудной клетки), интеллектуальный дефицит, вальгусная деформация нижних конечностей, рецидивирующие отиты, тугоухость и другие. При этом уровень ГАГ в моче в пределах нормы, отсутствует дефицит лизосомных ферментов. Молекулярно-генетическое исследование указывает на специфическую мутацию в гене AIFM1, что и позволяет установить верный диагноз.
Шаг 4. Дифдиагностика с заболеваниями, имеющими схожую с МПС клиническую симптоматику
И, наконец, необходимо исключить заболевания, под которые маскируются МПС: ювенильный ревматоидный артрит; детский церебральный паралич; гипертензионно-гидроцефальный синдром; периферическая нейропатия; гепатит неясного генеза; гепатолиенальный синдром; врожденный порок сердца; хронический отит, аденоидит, тонзиллит; хронический обструктивный бронхит; муковисцидоз; бронхиальная астма; гипотиреоз; иммунодефицитное состояние; помутнение роговицы.
Надо отметить, что часть этих диагнозов в контексте МПС лишь симптомы, например, помутнение роговицы, порок сердца, аденоидит или отит. Гепатит неясного генеза — симптом органомегалии, увеличения печени; муковисцидоз, хронический обструктивный бронхит — проявления поражения нижних дыхательных путей у пациентов с МПС.
Поэтому за постановкой любого диагноза должен стоять тщательный сбор анамнеза, включая неонатальный и семейный, а также объективная лабораторная диагностика. В случае с МПС этот принцип исключительно важен. Вот лишь пару примеров. Раньше детям с МПС часто выставляли детский церебральный паралич как окончательный диагноз. И это на фоне отсутствия отягощенного неонатального анамнеза, хронической гипоксии, повышения мышечного тонуса, позотонических и патологических рефлексов, характерных для ДЦП.
К диагнозам, которые прежде часто «подменяли» истинный, относится и гипотиреоз. Отчасти заболевание действительно напоминает МПС (крупный язык, задержка психоречевого развития, увеличенная щитовидная железа по данным УЗИ могут навести на мысль о болезни накопления). Однако у пациентов с МПС увеличение щитовидной железы обусловлено отложением ГАГ в тканях органа, которое при этом не дает никакого гормонального дисбаланса.
Читайте также: