Нейрома и нейрофиброма конъюнктивы: признаки, гистология, лечение, прогноз
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Каждому человеку хотя бы раз в жизни приходилось встречать людей, кожа которых покрыта пигментными пятнами и опухолеподобными разрастаниями. Скорее всего, вы увидели больного, страдающего нейрофибоматозом. Это одно из часто встречающихся наследственных заболеваний. Оно требует постоянного наблюдения и лечения у невролога, а также других специалистов. Иногда больному требуются проведение хирургических вмешательств.
Описание
Нейрофиброматоз - это заболевание относят к группе наследственных системных заболеваний. Оно принадлежит к группе факоматозов. При этих патологиях происходит сочетанное поражение кожи и структур нервной системы. Для него типичны нарушение развития эктодермальных и мезодермальных структур. К ним относят: кожу, нервную систему и скелет. Клинические проявления болезни могут появиться в любом возрасте.
Выделяют шесть типов нейрофиброматоза. Из них чаще встречаются и имеют большую клиническую значимость нейрофиброматоз I типа (болезнь Реклингхаузена), а также нейрофиброматоз II типа (нейрофиброматоз с невриномами VIII пары черепных нервов). Остальные формы встречаются довольно редко.
При этом заболевании образуются множественные нейрофибромы. Это опухоли доброкачественного характера. Они развиваются их клеток, составляющих оболочки нервных волокон. Эти образования чаще всего расположены в толще кожи и подкожной клетчатке, а также в межмышечном (мейсснеровом) и подслизистом (ауэрбаховом) пространстве.
Изредка они находятся в головном мозге, поражают нервные волокна, спинномозговые корешки, внутренние органы и мягкие ткани. На коже у больных появляются множественные пигментные пятна. У этих больных повышен риск развития злокачественных образований.
Причины
Нейрофиброматоз первого типа имеет наследственный характер, передается по аутосомно-доминантному типу наследования с высокой степенью пенетрантности. Оно одинаково часто встречается как у мужчин, так и у женщин. Ген НФ1, кодирующий заболевание, расположен в 17-й хромосоме на ее длинном плече. Он подавляет рост опухолевых тканей. При его дефекте отмечают нарушение синтеза белков, которые ответственны за размножение клеток.
Случаи заболевания нейрофибоматозом первого типа встречается примерно у одного ребенка на 3500 новорождённых. Эта патология обычно сочетается с развитием аномалий других органов и систем. Риск наследования при наличии болезни у одного из родителей составляет 50%. Если оно выявлено у обоих родителей, возможность рождения больного малыша составляет примерно 75%.
Причиной нейрофиброматоза второго типа считают мутацию гена НФ2, находящегося на 22 хромосоме. Он ответственен за продукцию белка мерлина, который является супрессором роста опухолей. Для этого заболевания характерен аутосомно-доминантный тип наследования с небольшой степенью пенетрантности. Другие типы нейрофиброматоза тоже связаны с мутациями в генах, кодирующих различные белки, регулирующие опухолевый рост.
Симптомы
Выраженность клинических проявлений при нейрофиброматозе обусловлен степенью поражения гена и недостаточностью выработки беков, подавляющих опухолевый рост. Самым характерным симптомом для нейрофиброматоза является гиперпигментация. У детей при рождении или несколько позже формируются пятна. Их цвет бывает разной интенсивности от светлого до темно-коричневого цвета, изредка они окрашены в багровый цвет.
Узелки Лиша (гамартомы) считают специфическим признаком нейрофиброматоза I типа. Их находят на радужке глаза. Они представляют собой пигментные пятна небольшого размера. Эти образования белесоватого или светло-бежевого оттенка. Они заметны только офтальмологу при специальном осмотре. Их становится больше по мере взросления человека.
В пубертатный период начинают появляться плексиформные и кожные нейрофибромы. Они располагаются под кожей, по ходу крупных нервов. Это мелкие доброкачественные опухоли, представляющие собой не более чем косметический дефект. Плексиформные образования, расположенные по длине периферических нервов, появляются на веках, конъюнктиве, в средостении, брюшной полости. Проявляются постоянной болью, онемением кожи, судорогами, параличами.
При этом заболевании опухоли ЦНС локализуются в полости черепа. Они представлены различными новообразованиями: глиомы зрительных нервов, астроцитомы, эпендимомы, невриномы слуховых нервов, менингиомы, нейрофибромы. Клинику определяет размер новообразований, вовлеченностью структур нервной системы в процесс. В детстве у этих больных выявляют расстройства развития: снижение интеллекта, гиперактивность.
При тяжелом течении болезни происходит деформация костей. У больного формируется сколиоз. Развиваются атрофии или гипертрофии трубчатых костей. Они искривляются, на поверхности их разрастаются гребни и наслоения. В костных полостях образуются нейрофибромы. При вовлечении в процесс костей черепа выявляют внешнюю асимметрию, которая выражена при поражении лица и глазниц. На своде черепа возникают участки атрофий, а изредка отмечают костные разрастания.
При нейрофиброматозе II типа образуются высокодифференцированные опухоли. Они ведут себя агрессивнее, чем при I типе заболевания.
Заметили у себя один или несколько симптомов?
Записаться на прием
Диагностика
Обследование больного для установления диагноза ведется группой специалистов. В нее входят: невролог, дерматовенеролог, офтальмолог, генетик, отоларинголог. У больного выясняют жалобы, его опрашивают с целью выяснения семейного анамнеза заболевания. При осмотре выявляют характерные поражения кожи, костей, нервной системы, внутренних органов.
Для уточнения диагноза проводят:
- МРТ, КТ. С их помощью выявляют наличие опухолей различной локализации. Находят деформацию костных структур, поражение внутренних органов.
- Рентген. Его проводят для выявления костных деформаций, разрастаний, эрозий.
- Офтальмологическое обследование. При его проведении выявляют специфические поражения для этого заболевания: гамартомы на радужке, нейрофибромы век и конъюнктивы, глиомы зрительных нервов.
- Исследование слуха. Его выполняют при наличии жалоб на усиливающуюся глухоту. Выявляют невриномы слуховых нервов.
Участки опухолей берут на биопсию. Затем проводится гистологическое исследование биоптата.
Лечение
Часто в ведении больных с нейрофиброматозом ограничиваются симптоматическим лечением. Если нейрофибромы расположены в местах повышенного травмирования и провоцируют боль, их удаляют хирургическими методами. При множественных образованиях назначают химиотерапию, проводят облучение. При поражении опорно-двигательного аппарата проводят реабилитационные мероприятия.
Заболевание имеет благоприятный прогноз. Опухолевые образования озлокачествляется крайне редко. При выполнении специальных мероприятий у больных сохраняется удовлетворительное качество жизни и работоспособность.
Врачи отделения
Табельский Роман Александрович
Врач невролог, врач нейрохирург, высшая врачебная квалификационная категория, стаж работы 17 лет
Нейрофиброматоз глаза и его придатков (болезнь Реклингаузена)
Что такое Нейрофиброматоз глаза и его придатков (болезнь Реклингаузена) -
Нейрофиброматоз глаза и его придатков - это местное проявление системного заболевания, имеющего наследственный характер.
Патогенез (что происходит?) во время Нейрофиброматоза глаза и его придатков (болезни Реклингаузена):
Этиология и патогенез неясны. Предполагают неправильное эмбриональное развитие мезенхимальных оболочек нервов. Нейрофиброматозные узлы обусловлены дисплазией нейроэктодермальной ткани. Узлы могут располагаться в глазу и его придатках. При нередкой внутричерепной локализации узлов могут возникать застойные диски зрительных нервов.
Симптомы Нейрофиброматоза глаза и его придатков (болезни Реклингаузена):
Для нейрофиброматоза характерны:
- множественные опухолевидные фиброматозные образования из оболочек нервов,
- пигментные пятна на коже цвета «кофе с молоком»,
- кожные и подкожные опухоли.
Нередко также отмечаются поражения костей (остеодистрофия), эндокринных желез, недоразвитие половой системы, умственная и физическая отсталость, реже нарушение психики. Нейрофиброматоз может сочетаться с пороками развития или другими опухолями (ангиомами, липомами). Чаще наблюдается у мужчин, обычно возникает в раннем детском возрасте.
Преимущественно встречается нейрофиброматоз век, почти всегда верхних. Веки утолщены, синюшного цвета, самостоятельно не поднимаются. Консистенция их тестовидная. Опухоль может распространиться на виски, лоб, проникать в орбиту, вызывать экзофтальм. При этом полость орбиты и канал зрительного нерва увеличены, обычно в вертикальном направлении. Костные стенки глазницы истончены, иногда узурированы. Могут поражаться и другие кости черепа. Нередко поражение век сочетается с гипертрофией соответствующей половины лица и с гидрофтальмом одноименного глаза. Болевых ощущений нейрофиброматоз обычно не вызывает.
Глазные симптомы встречаются почти у 20% больных. Разрастание узлов происходит по островкам цилиарных нервов, в результате чего появляются образования в области лимба, роговицы, радужки, сетчатки, зрительного нерва. В конъюнктиве склеры нейрофибромы имеют вид узелков, валикообразных утолщений. Узелки в радужной оболочке хорошо видны в свете щелевой лампы. Они, как правило, серого цвета, размерами не более просяного зерна, округлой формы, беспорядочно разбросаны по радужной оболочке и нередко имеются на обоих глазах.
При офтальмоскопии определяются изменения в сосудистой оболочке, сетчатой оболочке и на диске зрительного нерва в виде серовато-белых полосок. В позднем периоде преобладает застойный диск. Иногда эти изменения сочетаются с аномалиями развития лицевого и мозгового черепа, с гемигипертрофией лица, буфтальмом, гидрофтальмом, вторичной глаукомой. Как правило, поражениям придатков глаза и глазного яблока сопутствуют проявления нейрофиброматоза в других отделах лица и на туловище. Течение нейрофиброматоза длительное, прогрессирующее. Возможно злокачественное перерождение отдельных узлов в неврогенную саркому и появление метастазов.
Диагностика Нейрофиброматоза глаза и его придатков (болезни Реклингаузена):
Диагноз ставят на основании характерных клинических симптомов и данных рентгеновского исследования черепа и глазниц. Изменения в костях носят гиперпластический или атрофически-деструктивный характер. Для неврофиброматоза характерно увеличение глазницы и канала зрительного нерва в вертикальном направлении, что является дифференциально-диагностическим признаком при исключении других опухолей глазницы. Как правило, изменения в области глаза сочетаются с проявлением неврофиброматоза в других частях организма. Одновременно с неврофиброматозом наблюдается иногда и аномалии глаза: микрофтальм, микрокорнеа, вывих хрусталика. Нередко бывает глиома зрительного нерва.
Лечение Нейрофиброматоза глаза и его придатков (болезни Реклингаузена):
Лечение состоит в срочном удалении опухоли в пределах здоровой ткани, вплоть до экзентерации орбиты с помощью электроножа. В послеоперационном периоде проводится лучевая и химиотерапия с регулярными гемотрансфузиями.
Прогноз чаще неблагоприятный. Гибель около 80% больных наступает в течение 1-2 лет от генерализации процесса, метастазирования и кахексии.
К каким докторам следует обращаться если у Вас Нейрофиброматоз глаза и его придатков (болезнь Реклингаузена):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Нейрофиброматоза глаза и его придатков (болезни Реклингаузена), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Нейрофиброматозы
Нейрофиброматозы - наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое - проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10

Общие сведения
Нейрофибромы - доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз - болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом - 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.

Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых - нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) - супрессора опухолевого роста. Тип наследования - аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора - частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи - пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз - факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом - гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента - гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет - 41%, с 10 до 19 лет - 85%, после 20 лет - 95%.
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко - деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы - подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак - появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
- МРТ, КТ. Визуализационные методы исследований позволяют определить наличие нейрофибром в головном мозге, позвоночнике, внутренних органах. Двусторонние невриномы 7 пары черепных нервов являются диагностическим критерием нейрофиброматоза II типа. Часто выявляются глиомы, шванномы, менингиомы. Для I типа свойственно развитие плексиформных и обычных новообразований, глиом.
- Рентгенография костей скелета. Диагностическая процедура выполняется с целью подтверждения и оценки тяжести сколиоза, костных атрофий и гипертрофий, локальных утолщений и эрозийных поражений костных структур. При большинстве типов болезни наблюдается истончение кортикального слоя, ложные суставы, дисплазии крыльев клиновидной кости, дугообразное искривление большеберцовой и малоберцовой костей, кисты длинных костей.
- Офтальмологическое исследование. Нейрофиброматоз типа 1 сопровождается плексиформной нейрофибромой век, меланоцитарными гамартомами радужки, глиомой оптических нервных волокон, астроцитарной гамартомой сетчатки, утолщением роговичных нервов, конъюнктивальной нейрофибромой, ишемическими поражениями венул сетчатки. Патогномоничный признак - пятна светлого оттенка на глазном дне и радужке (гамартомы). При 2 типе диагностируется задняя субкапсулярная катаракта, помутнение хрусталика.
- Аудиологическое исследование. При опухолевом поражении слуховых нервов и жалобах на нарастающую глухоту (тугоухость) выполняется электрокохлеография и импедансометрия. Результаты указывают на снижение остроты слуха, наличие слуховой нейропатии, определяют причину и локализацию нарушения.
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями - малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
1. Нейрофиброматоз: этиология, патогенез, лечение/ Скаварская Е.А.// Международный журнал педиатрии, акушерства и гинекологии - 2014 - Т.5, №2.
2. Клинико-диагностические аспекты нейрофиброматоза/ Попова А.А.// Университетская медицина Урала - 2016 - №2.
3. Нейрофиброматоз первого типа (болезнь Реклингхаузена)/ Н.А. Шнайдер, А.И. Горелов// Сибирское медицинское обозрение - 2007.
Невринома ( Неврилеммома , Нейринома , Шванноглиома , Шваннома )
Невринома — это доброкачественное новообразование, возникающее из миелиновой оболочки нервного ствола. Становится причиной раздражения и дисфункции поражённого нерва, компрессии прилегающих тканей. Клинические проявления соответствуют расположению опухоли. Наиболее часто встречаются невриномы слухового нерва. Диагностика осуществляется комплексно по результатам неврологического осмотра, УЗИ, МРТ, КТ поражённой области, электронейромиографии, гистологического исследования. Лечение хирургическое, по показаниям проводится открытое или радиохирургическое удаление новообразования.

Причины невриномы
Шваннома образуется вследствие чрезмерного размножения шванновских клеток в неврилемме. Причины процесса усиленного деления остаются неизвестными. Значимыми факторами считаются:
- ионизирующая радиация
- плохая экологическая обстановка
- воздействие канцерогенов, поступающих в организм с пищевыми продуктами, вдыхаемым воздухом.
Предполагают наследственную детерминированность развития неврином. Склонность к их образованию отмечается у пациентов с нейрофиброматозом. Возникновение неврином глотки обусловлено хроническим вредоносным воздействием различных химических агентов, пыли, частых и длительных воспалительных процессов при хроническом тонзиллите, фарингите, назофарингите.
Макроскопически невринома является заключённым в капсулу, отграниченным округлым образованием с бугристой поверхностью. На разрезе опухоль имеет светло-серый или буро-коричневый цвет, определяются многочисленные фиброзированные участки, заполненные бурой жидкостью кисты. По мере роста неоплазия начинает сдавливать нервные волокна и окружающие нерв ткани, что вызывает возникновение основных клинических проявлений - нарушения функции нерва и прилегающих к нему структур. Выраженность симптоматики определяется локализацией шванномы. При расположении в узких рамках мосто-мозжечкового угла, костно-мышечного канала симптомы появляются рано, даже при небольших размерах образования.
Микроскопически невринома представляет собой параллельные ряды клеток с палочкообразными ядрами, перемежающиеся с волокнистыми структурами. Периферические участки опухоли окружены сосудистой сетью, центральные бедны сосудами. Вследствие недостаточного кровоснабжения в центральных отделах происходят дистрофические изменения. В результате последних невринома претерпевает различные морфологические трансформации, соответственно которым выделяют три основных гистологических типа неоплазии: эпителиоидный, ангиоматозный, ксантоматозный. Клинического значения указанная классификация не имеет.
Симптомы невриномы
Шваннома отличается медленным развитием симптоматики, может долго оставаться незамеченной. Признаки неоплазии зависят от ее месторасположения, включают две основные составляющие - симптомы расстройства функции поражённого нерва и проявления, обусловленные компрессией близлежащих тканей. При невриноме слухового нерва отмечается прогрессирующее снижение слуха. Поскольку опухоль односторонняя, пациент не сразу замечает развитие тугоухости. При вовлечении вестибулярной порции появляются головокружения с тошнотой и рвотой, вестибулярная атаксия.
Невринома тройничного нерва манифестирует прозопалгией, гипестезией половины лица, иногда - вкусовыми галлюцинациями. Поражение лицевого нерва характеризуется слабостью мимической мускулатуры. В ряде случаев тригеминальная и фациальная симптоматика провоцируются компрессией корешков соответствующих черепных нервов увеличивающейся вестибуло-кохлеарной шванномой.
Невринома спинального корешка протекает с классическим корешковым синдромом: болью, расстройством чувствительности, мышечной гипотонией, слабостью, атрофией в зоне иннервации поражённого корешка. Невринома периферического нерва проявляется аналогичными нарушениями в иннервируемой области. Со временем в денервированных тканях развиваются трофические расстройства. Невринома глотки вызывает чувство дискомфорта, обуславливает дисфагию, затруднение носового дыхания.
Неуклонно увеличивающаяся невринома мосто-мозжечкового угла может приводить к полной потере слуха, стойкому гемипарезу лица, внутричерепной гипертензии, сдавлению стволовых структур с развитием бульбарного синдрома, включающего нарушения глотания, речи, появление двоения в глазах. Компрессия мозжечка обуславливает возникновение мозжечковой атаксии: шаткость ходьбы, дискоординацию, крупноразмашистые движения, скандированную речь, нистагм. Спинальная невринома осложняется сдавлением спинного мозга с развитием компрессионной миелопатии, проявляющейся сенсомоторными нарушениями ниже очага поражения, расстройствами тазовых функций. Осложнением шванномы ветвей блуждающего нерва является нейропатический парез гортани.
Клиническая картина невриномы во многом сходна с поражением нервного ствола воспалительной, компрессионной, дисметаболической этиологии. Клиническая симптоматика, осмотр невролога позволяют определить уровень поражения. Последующая инструментальная диагностика направлена на уточнение морфологического субстрата, явившегося причиной поражения нервного ствола. Перечень необходимых обследований определяется расположением неоплазии, включает:
- Церебральную нейровизуализацию. Применяется при новообразованиях черепных нервов. Контрастная КТ головного мозга способна выявить невриномы размером более 1 см. Церебральная МРТ обладает большей информативностью, лучше визуализирует состояние окружающих опухоль тканей.
- МРТ. Проводится изолированная МРТ поражённого отдела позвоночника. Исследование позволяет обнаружить невриномы спинномозговых корешков, определить степень спинальной компрессии.
- Аудиометрия. Наряду с консультацией оториноларинголога и сурдолога, аудиометрия показана пациентам с понижением слуха. Обследование проводится для оценки степени тугоухости, исключения прочих возможных причин ухудшения слуха.
- КТ или МРТ гортани и глотки. Назначается при подозрении на невриному глотки. Выполняется после фарингоскопии для уточнения диагноза, локализации и размеров неоплазии.
- Сонография. УЗИ периферических нервов целесообразно при поражении нервных стволов конечностей. Дает возможность определить наличие локального утолщения неврилеммы. Более точно визуализировать образование помогает местная МРТ мягких тканей конечности.
- Электронейромиография. ЭНМГ необходима для анализа функционального состояния поражённого шванномой нервного ствола. В послеоперационном периоде применяется для контроля восстановления.
- Гистологическое исследование. Указанные выше обследования позволяют определить наличие опухолевого образования, предположить его доброкачественный характер. Точная верификация неоплазии возможна лишь по результатам исследования строения её тканей. Как правило, биопсия не назначается, проводится гистология операционного материала.
Дифференциальная диагностика шванном производится с другими опухолевыми образованиями. Невринома мосто-мозжечковой локализации требует дифференцировки от менингиомы, астроцитомы, опухоли мозжечка, спинальная шваннома — от иных экстрамедуллярных опухолей. Невриномы периферических нервов дифференцируют от компрессионно-ишемической, воспалительной невропатии.
Лечение невриномы
Единственным эффективным способом лечения шванном является их удаление. Выбор лечебной тактики осуществляется нейрохирургом, зависит от локализации неоплазии, возраста и состояния здоровья пациента. Применяются две основные методики:
- Хирургическое иссечение. Требует высвобождения нервных волокон от опухолевых тканей, что сопряжено с высоким риском травмирования, вероятностью сохранения отдельных опухолевых частиц, которые в последующем могут стать причиной рецидива. Для снижения риска указанных осложнений используется микрохирургическая техника. При интракраниальной локализации операция проводится путём трепанации черепа, при спинальной — с ламинэктомией.
- Радиохирургическое удаление. Выполняется при внутричерепной и спинальной локализации шванномы. Направленное лучевое воздействие вызывает гибель части опухолевых клеток, оставшиеся клетки утрачивают способность к размножению. Радикальная радиохирургическая операция возможна при размере неоплазии менее 30 мм, в остальных случаях радиохирургический метод применяется в качестве паллиативного лечения с целью уменьшения размеров новообразования у больных, имеющих противопоказания к открытой операции.
Исход заболевания зависит от расположения опухоли, своевременности диагностики и лечения. В большинстве случаев радикальное удаление обеспечивает благоприятный результат. В отдельных случаях наблюдается рецидив новообразования. Отсутствие лечения приводит к необратимой потере функции поражённого нервного ствола, возникновению осложнений. Специфическая профилактика не разработана, общие профилактические меры сводятся к предупреждению воздействия онкогенных факторов, повышению противоопухолевого иммунитета.
1. Магнитно-резонансная томография в нейрохирургии/ Коновалов А.Н., Корниенко В.Н., Пронин И.Н. - 1997.
3. Невриномы нервных стволов конечностей: клиника, диагностика и лечение/ Цымбалюк В.И., Третяк И.Б., Тончев М.Д.// Украинский нейрохирургический журнал. - 2008.
Нейрофиброматоз: отражение проблемы в глазах
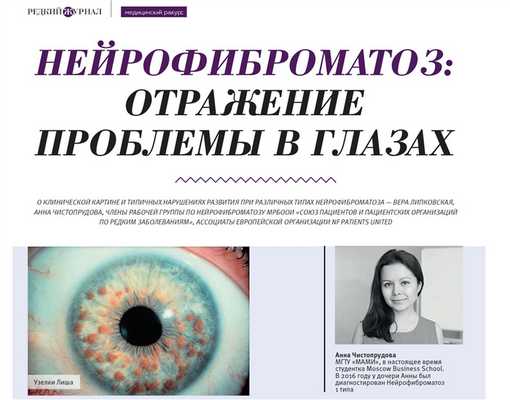
О клинической картине и типичных нарушениях развития при различных типах нейрофиброматоза — Вера Липковская, Анна Чистопрудова, члены рабочей группы по нейрофиброматозу мрбоои «Союз пациентов и пациентских организаций по редким заболеваниям», ассоциаты европейской организации NF PATIENTS UNITED.
ГЕНЕТИЧЕСКОЕ ОТКЛОНЕНИЕ
Нейрофиброматоз, также известный как болезнь (синдром) Реклингхаузена, представляет собой генетическое заболевание, которое, по разным
данным, наблюдается примерно у 1 из 2500-3500 новорожденных детей. Возникновение болезни может быть обусловлено наследственными факторами,
а в ряде случаев — спонтанной мутацией генов. Гены, ответственные за возникновение нейрофиброматоза, локализованы на длинном плече 17 хромосомы 17q11.2, в которой содержится информация, ответственная за синтез нейрофибромина и других белков. Ген НФ-1 выступает как супрессор опухолевых
процессов, а потому в результате мутации нарушается общий иммунитет к
возникновению опухолей, которые в большинстве случаев являются доброкачественными.
Первые упоминания о данной болезни встречаются в литературе XIX в.,
когда ирландским хирургом Робертом Смитом в отдельную группу были выделены пациенты с опухолями на нервных тканях. Впоследствии заболевание
было описано в 1882 году в монографии немецкого патологоанатома Фридриха фон Реклингхаузена, который описал клиническую картину и патологические основы появления нейрофибром (доброкачественных опухолевых образований). В 1937 году австрийский офтальмолог Карл Лиш описал узелки
Лиша (Lisch nodules) у пациентов с нейрофиброматозом. В 1956 году исследователь Франк Кроуэ и его коллеги впервые определили, что данное заболевание является наследственным и передается потомству в
50% случаев. В 1988 году Национальные институты здравоохранения США (National In itutes of Health Consensus
Development) провели первую конференцию по нейрофиброматозу с целью выработки последовательных критериев его диагностики. По итогам конференции был принят
доклад, в котором выделялось 7 типов нейрофиброматоза.
3 ТИПА ЗАБОЛЕВАНИЯ
В настоящее время в зарубежной практике принято
выделять 3 типа заболевания: нейрофиброматоз I типа, нейрофиброматоз II типа и шванноматоз.
Для удобства нейрофиброматоз I типа и нейрофиброматоз II типа далее также будут сокращенно именоваться НФ1 и НФ2 соответственно.
Диагноз нейрофиброматоз I типа может быть поставлен
при наличии сочетания двух и более симптомов:
- шесть и более пятен цвета «кофе с молоком», которые
появляются в течение первых 2 лет жизни;
- наличие двух и более обычных нейрофибром, либо одной плексиформной нейрофибромы;
- гиперпигментация в подмышечной и/или паховой
- глиомы зрительных нервов;
- два и более узелка Лиша;
- ложный сустав и другие костные патологии;
- наличие нейрофиброматоза I типа у ближайших родственников.
Указанные симптомы проявляются у пациентов в различные периоды жизни и практически на всем ее протяжении. Часть из них формируется в течение первых двух
лет жизни, другие к 5-7 годам, и т.д.
Помимо собственно клинической картины заболевания, оно также может стать причиной задержки развития, когнитивных нарушений, проблем психосоциального характера, нарушения координации и т.д.
Выделяют следующие основные группы нарушений в развитии при НФ1, часто являющиеся причиной низкой успеваемости при обучении: моторные навыки; речевые навыки; письмо; память, внимание и мышление;
визуально-пространственное восприятие; проблемы с планированием и организацией; проблемы с поведением и социальным взаимодействием.
Во многих случаях у пациентов с НФ1 наблюдаются расстройства аутистического спектра (РАС) и синдром дефицита внимания и гиперактивности (СДВГ), депрессивные и тревожные состояния.
Нейрофиброматоз — не приговор, и случается, что люди с данным заболеванием живут, совершенно не подозревая о его наличии, пока не проявится какой-либо из клинических симптомов, иногда уже во взрослом возрасте. Вместе с тем, многие пациенты постоянно находятся в зоне риска, поскольку заболевание неизлечимо и может дать осложнение в любой момент. Именно поэтому вопрос своевременной диагностики и доступного медицинского обслуживания стоит особенно остро, так
как пациенты с НФ1 должны постоянно наблюдаться у дерматолога, невролога, ортопеда, офтальмолога, кардиолога, онколога, психолога.
К сожалению, сегодня пациентам с НФ1 в России, особенно, в регионах, не так просто получить качественное медицинское обслуживание в силу того, что не все врачи
имеют большой опыт и знания в области данного заболевания. Понимание особенностей клинической картины НФ1 и дальнейшее изучение и обобщение опыта в этой сфере поможет обеспечить людям, страдающим данным заболеванием, более качественную поддержку и улучшить тем самым качество их жизни.
Нейрофиброматоз II типа встречается реже, примерно у 1 из 50 000 новорожденных. Молекулярно-генетические исследования выявили принципиальные отличия в патогенезе НФ1 и НФ2, которые представляют собой разные заболевания и
требуют дифференцированного клинического подхода. НФ2 формально является аутосомно-доминантным генетическим заболеванием. Возникающие при НФ2 опухоли являются доброкачественными, но биологически более агрессивными, чем при НФ1.
Диагноз нейрофиброматоз II типа может быть поставлен при наличии сочетания двух и более симптомов:
- опухоли восьмого черепно-мозгового нерва (т.н. невриномы или шванномы);
- менингиома или иные опухоли мозга;
- звон в ушах (тиннит), утрата слуха и/или глухота;
- появление катаракт в раннем возрасте;
- опухоли спинного мозга;
- нарушение равновесия;
- атрофия мышц.
При НФ2 происходит развитие опухолей на восьмом черепно-мозговом нерве и вестибулярных нервах, что зачастую вызывает давление на слуховые нервы и приводит к потере слуха, и данная угроза сохраняется на протяжении всей жизни. Также примерно в подростковом возрасте появляются: звон в ушах, онемение лица,
головокружение, нарушение баланса тела, хронические головные боли. При наличии опухолей спинного мозга возможно онемение других частей тела.
В течение последних нескольких лет зарубежные ученые ведут дискуссию о пересмотре диагностических критериев НФ2. В частности, в очередной раз намерение
по пересмотру озвучивалось его инициаторами в рамках Международной конференции по нейрофиброматозу, прошедшей осенью 2018 года в Париже (Франция). Шванноматоз, также иногда именуемый нейрофиброматоз III типа, представляет собой крайне редкое генетическое заболевание, встречающееся с частотой примерно 1 случай на 1,7 млн человек. Впервые данное заболевание было описано
у пациентов из Японии, у которых наблюдались множественные кожные шванномы, опухоли ЦНС и другие неврологические осложнения, однако в отсутствие типичных для НФ клинических симптомов.
В основном, типичные для НФ2 и шванноматоза доброкачественные опухоли вырастают из т.н. шванновских клеток — глиальных клеток, образующих миелиновую оболочку нервов. В случае, когда шванновские клетки начинают бесконтрольно распространяться, они образуют своего рода капсулу, которая называется шванномой.
Несмотря на то, что сами по себе шванномы являются доброкачественными, их наличие может стать опасным в случаях, когда их разрастание приводит к сдавливанию нервов, возникновению хронических болей, что зачастую обусловливает
необходимость хирургического вмешательства или применения других методов
лечения. В частности, хронические боли у пациентов с шванноматозом являются предметом углубленного изучения в зарубежной науке, поскольку их наличие
Читайте также:
- Синдром Дюпре (Dupre)
- Генерализованное поражение клеток при шоке. Ацидоз при шоке и некрозы тканей
- Этмологическая психиатрия. История этмологической психиатрии
- Как избавиться от глазного клеща? Причины и симптомы заражения демодексом
- Выявление противовирусных антител ( AT ) в сыворотке крови. РТГА. РСК. РИФ. Иммуносорбционные методы выявления противовирусных антител.