Порокератозы у ребенка (Мибелли, линейный, диссеминированный, точечный)
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Болезнь Кирле (кератоз фолликулярный и парафолликулярный, проникающий): клинический случай
Журнал: Клиническая дерматология и венерология. 2011;9(5): 25‑28
Карпов В.В. Болезнь Кирле (кератоз фолликулярный и парафолликулярный, проникающий): клинический случай. Клиническая дерматология и венерология. 2011;9(5):25‑28.
Karpov VV. Kyrle's disease (follicular and parafollicular, penetrating keratosis): a case report. Klinicheskaya Dermatologiya i Venerologiya. 2011;9(5):25‑28. (In Russ.).
Рассмотрены вопросы эпидемиологии, этиопатогенеза, клинические проявления, методы диагностики и лечения парафолликулярного проникающего кератоза (болезни Кирле). Представлено собственное клиническое наблюдение.
Кератоз фолликулярный (парафолликулярный кератоз, болезнь Кирле — БК) — редкое заболевание кожи из группы перфорирующих дерматозов, в основе которого лежит нарушение ороговения с интрадермальной инвагинацией эпидермиса [1] (синонимы: гиперкератоз фолликулярный и парафолликулярный, проникающий в кожу, гиперкератоз фолликулярный и парафолликулярный с пенетрацией в кожу, (hyperkeratosis follicularis et parafollicularis in cutem penetrans), проникающий гиперкератоз [2, 3]).
Заболевание в 1916 г. впервые описал австрийский дерматолог Дж. Кирле (J. Kyrle), наблюдавший этот дерматоз у больного сахарным диабетом [3—5].
Эпидемиология. Заболевание редкое. Возраст больных 20—70 лет. Женщины болеют чаще, дети — очень редко. Сопутствующие заболевания: часто — сахарный диабет и хроническая почечная недостаточность, реже — сердечная недостаточность, гипотиреоз, гиперлипопротеинемия II типа (повышение в крови уровня холестерина и триглицеридов) [1, 3—6].
Этиология и патогенез окончательно не изучены. Ранее предполагаемая роль вирусной или бактериальной инфекции, нарушение обмена витамина А в развитии этого дерматоза представляет лишь исторический интерес [4]. Определенное значение в развитии заболевания, помимо нарушения углеводного обмена, придается поражению печени (хронический гепатит) с развитием вторичной недостаточности витамина А [5].
Генетическая предрасположенность к развитию БК окончательно не доказана, хотя описаны случаи заболевания среди родственников в одной семье при кровном родстве (родственники первой степени) [1, 3—5].
По первоначальному определению Дж. Кирле [7], это заболевание, при которой атипичный клон кератиноцитов проникает через эпидермис в дерму. Полагают, что в основе патологического процесса лежит нарушение кератинизации, дифференцировки и ороговения кератиноцитов (образование дискератотических фокусов и ускорение процесса ороговения). Это ведет к образованию кератотических пробок с участками паракератоза. Ороговение начинается уже на границе эпидермиса и дермы. Скорость дифференцировки и кератинизации превышает скорость клеточной пролиферации, поэтому паракератотический конус частично проникает глубже, в нарушенный эпидермис, и обусловливает перфорацию его в дерму [1, 3—6].
Единой классификации фолликулярных кератозов нет. Среди самостоятельных нозологических форм выделяют папулезные, атрофирующие и вегетирующие фолликулярные кератозы [8]. Ряд авторов [4, 6] обращают внимание на сходство БК с лентикулярным стойким гиперкератозом Флегеля и рассматривают последний как вариант БК, хотя клинические проявления различаются. Другие авторы [5, 8] относят БК к вегетирующим фолликулярным кератозам, а кератоз фолликулярный стойкий (болезнь Флегеля) — к папулезным.
Рассматривается концепция реактивного перфоративного нарушения кератинизации на фоне болезней почек, печени, сахарного диабета. Появилось понятие — перфорирующие заболевания кожи, к нему относят группу заболеваний, при которых происходит элиминация измененных компонентов кожи через эпидермис (процесс, называемый трансэпидермальной элиминацией). Некоторые авторы оспоривают БК, как самостоятельную нозологическую единицу [7].
В Международной классификации болезней 10-го пересмотра в классе XII (болезни кожи и подкожной клетчатки) выделена рубрика L87 — трансэпидермальные прободные изменения, в которой БК отведена отдельная подрубрика L87.0.
Патоморфология весьма характерна и имеет диагностическую значимость. Углубления эпидермиса и расширенные устья волосяных фолликулов заполнены гиперкератотическими пробками. Под пробками в эпидермисе выражено разрастание зернистого слоя, а в местах отсутствия гипергранулеза — паракератоз, проникающий весь эпидермис до дермы. В дальнейшем происходит истончение эпидермиса и проникновение в дерму роговых масс с формированием в дерме воспалительных инфильтратов типа гранулем из лимфоцитов, лейкоцитов, гистиоцитов и гигантских клеток инородных тел. Наблюдается гибель сальных желез в области пораженных фолликулов, дистрофия коллагеновых волокон, гиперэластоз [1, 2, 5, 6].
Клиническая картина. Начало заболевания постепенное, новые высыпания появляются по мере исчезновения старых элементов. Характерны фолликулярные или парафолликулярные папулы сначала цвета здоровой кожи, а впоследствии сероватой или буровато-красной окраски, диаметром от булавочной головки до 1 см. В центре элементов — роговая пробка, при удалении которой образуется кратерообразное углубление. Папулы склонны к периферическому росту и слиянию с формированием сухих полициклических бляшек, покрытых наслоением чешуек, корочек. Консистенция плотная, поверхность неровная, бородавчатая. Свежие высыпания сопровождаются легким зудом (чаще у больных сахарным диабетом) или не беспокоят больного. Старые, более крупные очаги бывают болезненными, особенно при надавливании. В местах расчетов линейное расположение, что свидетельствует о возможности возникновения феномена Кебнера. Возможно присоединение вторичной инфекции. Локализация высыпаний — разгибательные поверхности конечностей, туловище, ягодицы. Слизистые оболочки не поражаются [1—6]. Высыпания в полости рта, на ладонях, подошвах, гениталиях встречаются редко [4].
Течение БК хроническое, рецидивирующее. Заболевание с трудом поддается лечению. Прогноз во многом зависит от основного заболевания.
Диагностика основывается на анамнезе, клинической и гистологической картине заболевания. Дифференциальную диагностику БК необходимо проводить с красным волосяным лишаем Девержи, фолликулярным дискератозом Дарье, порокератозом Мибелли, трансэпидермальными прободными изменениями кожи (перфорирующий ползучий/серпигинозный эластоз, реактивный перфорирующий коллагеноз), болезнью Флегеля [8].
Болезнь Флегеля (hyperkeratosis lenticularis perstans), так же как и БК, является редкой формой фолликулярного кератоза. Предполагают, что в основе данного заболевания лежит наследственно обусловленная (аутосомно-доминантный тип) патология ороговения, выражающаяся в нарушении синтеза кератина 55К, содержание которого резко снижено в очагах поражения при болезни Флегеля, в то время как в нормальной коже этот вид кератина доминирует. Гистологически выявляют истончение эпидермиса, фолликулярный ортокератоз с фокусами паракератоза, местами спонгиоз, в дерме выражен полосовидный лимфоцитарный (преимущественно Т-хелперный) инфильтрат. Придававшееся ранее большое значение в патогенезе дерматоза отсутствию или нарушению структуры телец Одланда [4] в настоящее время подвергается сомнению. Гранулы Одланда появляются в верхних рядах шиповатого слоя в кератиноцитах, занимают периферическое положение и выделяют путем экзоцитоза свое содержимое в межклеточное пространство, где оно принимает пластинчатое строение (межклеточный цемент) — компонент нормальной кератинизации. В отличие от БК этот дерматоз проявляется чаще в юношеском и среднем возрасте, характеризуется образованием диссеминированно расположенных более мелких (1-5 мм) и более поверхностно залегающих роговых папул (без тенденции образовывать бляшки). При БК нет нарушений кератина 55К [6].
Лечение: витамин А, аевит длительно, ретиноиды (неотигазон, этретинат, ацитретин из расчета 0,5 мг/кг в сутки). Наружно - кератолитические мази, 2% серносалициловая мазь, ретиноидный крем, 0,1% тигазоновый крем, 5% фторурациловый крем (на площадь не более 500 см одновременно), электро-, крио- и лазеродеструкция (углекислый лазер) отдельных очагов, ультрафиолетовое облучение. Эффект от лечения временный. После удаления роговой пробки остается атрофический, неравномерно пигментированный рубец; рецидив может возникнуть рядом. Необходима коррекция висцеральной патологии [3—6].
Приводим собственное клиническое наблюдение.
Больной Б., 57 лет, пенсионер (музыкант военного оркестра), поступил в отделение гнойной хирургии 13.09.10 на оперативное лечение по поводу остеомиелита левой стопы. Лечащим врачом назначена консультация дерматолога из-за высыпаний на коже.
Анамнез. Первые высыпания на коже грудной клетки спереди появились около 20 лет назад. Высыпания практически не беспокоили, редко отмечался незначительный зуд, больше «волновал» косметический аспект. Выставлялись диагнозы: дерматит, ксероз, кератоз. Эффекта от рекомендованных салициловой мази, кремов с ромашкой, чередой, приема витамина А внутрь не отмечалось, что объясняли «своеобразным строением кожи пациента». По словам пациента, наилучший эффект отмечал от применения циндола («болтушка хорошо подсушивает и слущивает пробочки» на коже). Полностью высыпания не исчезали, сезонность не отмечена.
С 1995 г. страдает сахарным диабетом 2-го типа, принимал перорально гипогликемические препараты (диабетон). В 2009 г. стационарно лечился по месту жительства по поводу синдрома диабетической стопы, гнойно-некротической раны на левой стопе; заживление вторичным натяжением. С лета 2010 г. высыпаний стало значительно больше, что пациент связывал с сильной жарой. В июле получил колотую рану левой стопы (наступил на гвоздь на даче), которая длительно не заживала, повысился уровень сахара крови. В связи с ухудшением состояния выраженного болевого синдрома пациент госпитализирован.
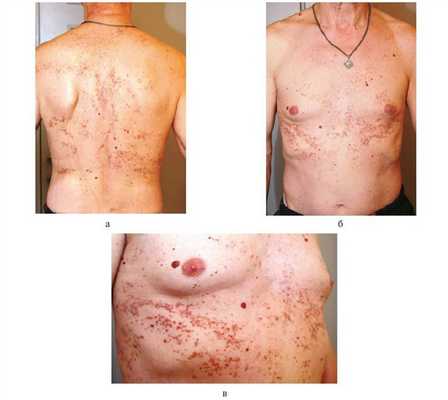
Кожный статус. На коже туловища — множественные фолликулярные и парафолликулярные папулы красно-бурого цвета, местами сгруппированные в мелкие бляшки, покрытые плотными чешуйками. В центре папул — роговые пробки. Там, где пробки отторглись, кратерообразные углубления, заполненные нанесенным ранее циндолом. На месте разрешившихся элементов — мелкие рубчики. При пальпации высыпания плотные, кожа напоминает «терку», влажная. Несколько внутридермальных невусов округлой куполообразной формы, мягкой консистенции, коричневого цвета (рисунок, а—в). Рисунок 1. Пациент Б., 57 лет: проявления БК. Пациент страдает сахарным диабетом, диабетической нефропатией. а—в — множество розовато-красного и сероватого цвета папул и узлов с роговыми пробками. В тех местах, где больной расчесывал кожу, элементы сыпи располагаются линейно (симптом Кебнера). Несколько внутридермальных невусов. Кожа головы, конечностей, видимые слизистые оболочки без высыпаний, физиологической окраски.
Результаты специальных обследований. Анализ крови клинический: гемоглобин — 124 г/л, эритроциты — 4,0·1012/л, лейкоциты — 6,7·109/л (при поступлении 12,8·109/л), нейтрофилы — 67%, сегментоядерные — 65%, палочкоядерные — 2%, эозинофилы — 3%, лимфоциты — 23%, моноциты — 9%, скорость оседания эритроцитов — 35 мм/ч (при поступлении 53 мм/ч). Титр АСТЛ-0 и ревматоидный фактор — не обнаружены. СРП —10 мг/л. Анализ мочи при поступлении: удельный вес — 1020, белок — 0,132‰, сахар — 0,3 мг/л, ацетон — нет, лейкоциты — 3—5 в поле зрения, эритроциты — 4—5 в поле зрения, цилиндры — 4 в препарате. При выписке анализ мочи в норме.
Рентгенография органов грудной клетки — без патологии. На рентгенограммах левой стопы - полная деструкция головки III плюсневой кости, частичное разрушение II плюсневого сустава. Электрокардиограмма — ритм синусовый, частота сердечных сокращений 76 в минуту; изменения в миокарде; феномен ранней реполяризации желудочков.
Эндокринолог. Сахарный диабет 2-го типа, тяжелое течение, инсулинопотребный, декомпенсированный. Диабетическая ангиопатия сосудов нижних конечностей. Диабетическая нефропатия. ХПН-0.
Офтальмолог. Пролиферативная диабетическая ретинопатия. Начальная катаракта правого глаза.
Хирург. Вялогранулирующие раны подошвенной поверхности левой стопы. Остеомиелит II—III плюсневых костей левой стопы.
Дерматолог. Кератоз фолликулярный и парафолликулярный, проникающий (БК). Внутридермальные невусы. Противопоказаний к оперативному лечению нет.
Больному 20.09.10 произведена трансметатарзальная резекция головки III плюсневой кости. Ампутация III пальца левой стопы. Получал антибактериальную терапию, хумулин, лантус; проводились перевязки с димексидом, мазями на водорастворимой основе, физиотерапия. Кожу туловища 1 раз в день обрабатывал циндолом.
Рекомендации при выписке. Наблюдение эндокринолога, хирурга по месту жительства; хумулин по 18 ЕД 3 раза в день перед основным приемом пищи, лантус в 22:00 по 16 ЕД. Перевязки остаточных ран с мазью бетадин. Лазерная коагуляция сетчатки после нормализации сахаров в отдельную госпитализацию, тогда же показаться дерматологу.
Каждый дерматолог встречается с непонятными больными. Особенно это касается редких дерматозов, с некоторыми из них врач может столкнуться лишь раз за свою практику. «Лучше один раз увидеть, чем сто раз услышать», — гласит русская народная мудрость. «Главный инструмент дерматолога — его глаза» [3].
То что у нашего пациента выраженные явления гиперкератоза туловища, было очевидно, но мы не встречались раньше ни с БК, ни с болезнью Флегеля. Только на основании анамнестических данных, характерной клинической картины и сведений, полученных из источников литературы (от предложенной биопсии кожи больной отказался), был выставлен окончательный диагноз. Наш пациент страдает сахарным диабетом, диабетической нефропатией. Если бы рекомендуемое обследование всех больных с гиперкератозами для выявления латентного диабета [4] было сделано раньше, возможно, и диагнозы были выставлены раньше. Цель данной публикации заключается именно в этом.
Порокератозы у ребенка (Мибелли, линейный, диссеминированный, точечный)
Заболевание проявляется у взрослых, обычно в возрасте от 30 до 60 лет, без различия пола, только у представителей белой расы. Часто наблюдаются семейные формы, указывающие на аутосомно-доминантный характер наследования.
Элементы многочисленные, локализуются на открытых участках кожного покрова, особенно на разгибательных поверхностях предплечий и голеней, симметрично. Лицо, шея, верхняя часть груди, плечи и тыльная поверхность стоп поражаются в меньшей степени. Первичным элементом является мелкая кератотическая папула, которая уплощается, увеличивается в размере и превращается в пятно розовато-коричневого цвета диаметром 5-10 мм, плоское или слегка атрофичное . При рассматривании элементов под лупой у большинства из них обнаруживается очень тонкий роговой бортик более темного цвета, окаймляющий пятно. При пальпации определяется легкая шероховатость.
Диагностика основывается на характерной клинической картине и данных гистопатологического исследования.Основным гистологическим признаком актинического порокератоза является наличие роговидной пластинки, представляющей собой столбик из паракератических клеток, в заполненном роговыми массами углублении эпидермиса. Углубление может располагаться как в устье потовой железы, так и в устье волосяного фолликула. Под столбиком отсутствует зернистый слой, встречаются вакуолизированные дискератические клетки. В эпидермисе вокруг роговидной пластинки отмечаются акантоз, папилломатоз и гиперкератоз, а также истончение мальпигиева слоя и поверхностный полосовидный инфильтрат с базофильной дистрофией коллагена.
- Дифференцируют от порокератоза Мибелли, который возникает в детском возрасте и образует одиночную бляшку неправильных очертаний, диаметром до 10 см, чаще на тыльной поверхности кисти. Центр бляшки слегка западает, атрофичен, окружен валиком, по краю которого проходит бороздка с вертикально поднимающимся роговым гребнем. Реже встречается линейное (зостериформное) расположение элементов, преимущественно локализованных на разгибательных поверхностях конечностей, на кистях и стопах. Морфологическая картина при порокератозе Мибелли идентична актиническому порокератозу.
Эффективных методов лечения не существует.
Нижеследующий методы лечения применяются с переменным успехом:
Уильямс Г.М., департамент здравоохранения Сан-Антонио; Филлман Э.П., Военно-медицинский центр Сан-Антонио
Введение
Порокератоз - редкое дерматологическое заболевание. Это нарушение кератинизации, которое клинически представляет собой кератотические папулы или кольцевые бляшки с приподнятыми краями (1).
Он имеет отчетливый гистологический признак - наличие роговой пластинки, которая представляет собой колонну плотно прилегающих паракератотических клеток в верхней части эпидермиса (2, 3).
Существует несколько различных клинических вариантов порокератоза, в том числе диссеминированный поверхностный актинический порокератоз, порокератоз Мибелли, ладонно-подошвенный порокератоз и диссеминированный, а также линейный порокератоз. Кроме того, более редкие варианты включают генитальный порокератоз, лицевой порокератоз, гигантский порокератоз, птихотропный порокератоз, гипертрофический веррукозный порокератоз, эруптивный зудящий папулезный порокератоз, фолликулярный порокератоз и ретикулярный порокератоз (1, 4).
Эти варианты могут встречаться одновременно (5), но это редкий случай (1). Порокератоз - э о предраковое заболевание, которое может подвергнуться злокачественной трансформации (6).
Оценку порокератоза лучше всего проводить при биопсии приподнятой периферической части элемента. Хотя существует несколько методов лечения порокератоза, включая местную, системную и хирургическую терапию, стандартных рекомендаций по лечению не существует (7).
Этиология
Этиология порокератоза в настоящее время неясна (3).
Эпидемиология
Порокератоз - редкий диагноз. Обычно поражения появляются на открытой солнцу коже, чаще всего на пятом десятилетии жизни, но может происходить в любом возрасте и с одинаковой частотой у мужчин и женщин (2). Повышенная распространенность диссеминированного поверхностного актинического порокератоза на открытой солнцу коже, вероятно, указывает на то, что ультрафиолетовое излучение является фактором риска (8).
Эруптивная форма порокератоза коррелирует с иммуносупрессией, трансплантационными операциями, воспалительными состояниями и злокачественными новообразованиями (4). Порокератоз прогрессирует в немеланомный рак кожи в 6,9-30% случаев, чаще всего плоскоклеточный рак и реже базальноклеточный рак (6).
Хотя порокератоз обычно является приобретенным заболеванием, часто существует семейная предрасположенность, которая может означать наличие наследственного компонента (5).
Патофизиология
Порокератоз - это явление, возникающее в результате нарушения дифференцировки клеток эпидермиса (9). Развитие этого состояния может быть связано с воздействием солнца. В местах, где воздействия солнца не было или было ограничено пребывание на солнце, повторная незначительная фрикционная травма из-за тесной одежды может вызвать образование элементов сыпи, как это имеет место при урогенитальном порокератозе (1).
Сообщается об ассоциации с избыточной экспрессией р53, а иногда может наблюдаться экспансия клона аномальных эпидермальных кератиноцитов (6, 5). Порокератоз имеет возможность злокачественной трансформации в плоскоклеточный рак или базальноклеточный рак (5).
Гистопатология
Отличительной гистопатологической особенностью порокератоза является роговая пластинка, представляющая собой колонну плотно прилегающих паракератотических клеток (2, 3). Столбик паракератоза возникает над эпидермисом с отсутствующим зернистым слоем и дискератотическими клетками в верхней части шиповатого слоя (1).
Эта особенность обычно присутствует на приподнятом крае очага поражения (1). Хотя когда-то этот признак считался патогномоничным для порокератозом, эта особенность проявилась и при других заболеваниях (9). Роговая пластинка отражает неправильную дифференцировку клеток эпидермиса (9). При фолликулярном порокератозе роговая пластинка может вовлекать фолликулярный инфундибулум (10).
Клиника и физикальное обследование
Порокератоз проявляется в виде кератотических папул или кольцевых бляшек, склонными к периферическому росту с приподнятой кератотической периферией (1). В центре очаг поражения может казаться слегка атрофичным (1). Эти поражения могут сопровождаться зудом и могут присутствовать в течение нескольких лет до постановки диагноза (1).
Диссеминированные узелки представлены в виде розовых или коричневых папул и пятен с приподнятыми границами (4). Порокератоз чаще всего возникает в конечностях и туловище, но может возникать на лице, области половых органов и мошонки (10). Однако порокератоз как синдром имеет широкий спектр проявлений, включая значительные деструктивные поражения и формирование рубцов (6).
Обследование
Обследование пациентов с порокератозом лучше всего проводить методом биопсии края очага поражения (1). Биопсия из этой области выявляет роговую пластинку и будет по существу являться диагностической. Дерматоскопия также может быть полезна для диагностики. Дермоскопия очагов поражения выявляет центральную коричневую пигментацию с сине-серыми точками, окруженными единственной гипопигментированной полосой с белым ободком по периферии (1).
Несмотря на относительную редкость порокератоза, особенно некоторых подтипов, таких как порокератоз урогенитальной локализации, следует иметь в виду такой диагноз из-за риска злокачественной трансформации (6).
Лечение / Ведение пациентов
- Описаны различные методы лечения порокератоза, включая местную, системную и хирургическую. Однако рандомизированных контролируемых исследований не проводилось, поэтому не существует международных клинических рекомендаций по стандартам лечения (7).
- Классический порокератоз Мибелли наиболее успешно лечится кремом с имиквимодом (7).
- Линейный порокератоз хорошо реагирует на местные или системные ретиноиды (7).
- Диссеминированный порокератоз можно успешно лечить местными производными витамина D (7).
- Хирургические вмешательства или криотерапия - это варианты лечения поражений в тех областях, где применение местных препаратов затруднено или противопоказано (7). Лазеротерапия может быть еще одним вариантом лечения (4).
- Местные стероиды, ретиноиды и местное применение диклофенака могут обеспечить симптоматическое лечение, даже если стойкого эффекта не наблюдается (1).
Дифференциальная диагностика
Клинически порокератоз может напоминать псориаз, простой хронический лишай, гипертрофический плоский лишай, туберкулез кожи, болезнь Боуэна, кандидоз, раздражительный и аллергический контактный дерматит (1).
Прогноз
Порокератоз является предраковым состоянием (1); однако все виды порокератоза могут подвергаться злокачественной трансформации в немеланомный рак кожи с вероятностью от 6,9 до 30% (2, 6). Чаще всего заболевание трансформируется в плоскоклеточный рак. Реже заболевание трансформируется в базальноклеточный рак. Если злокачественной трансформации не произошло, иссечение является методом выбора.
Линейный порокератоз и гигантский порокератоз являются вариантами, наиболее подверженными злокачественной трансформации. Злокачественная трансформация наиболее редка при диссеминированном поверхностном актиническом порокератозе (11).
Информирование и обучение пациентов
Пациенты должны пользоваться средствами защиты от солнца, избегать чрезмерного воздействия солнечного света и периодически проходить обследование у дерматолога для контроля рецидива или злокачественной трансформации (2).
Другие аспекты
- Гистологическим признаком порокератоза является роговая пластинка, представляющая собой колонну плотно прилегающих паракератотических клеток (3).
- Порокератоз проявляется в виде кератотических папул или кольцевидных бляшек, которые имеют тенденцию к периферическому росту с приподнятым кератотическим краем (1). Дермоскопия очагов поражения выявляет центральную коричневую пигментацию с сине-серыми точками, окруженными единственной гипопигментированной полосой с белым венчиком по периферии (1).
- Существует множество вариантов порокератоза (2).
- Порокератоз может подвергаться злокачественной трансформации в плоскоклеточный рак и базальноклеточный рак (6).
- Оценку порокератоза лучше всего проводить с помощью биопсии на границе очага (1).
- Описаны различные методики лечения порокератоза, включая местную, системную и хирургическую. Однако рандомизированных контролируемых исследований не проводилось, поэтому не существует международных клинических по стандартам лечения (7).
- Пациентам, получающим лечение от порокератоза, следует рекомендовать использовать солнцезащитные средства, избегать чрезмерного воздействия солнечных лучей и периодически проходить обследование у дерматолога для контроля рецидива или злокачественной трансформации (2).
Улучшение результатов работы медицинского персонала
Дерматологи и другие клиницисты должны тесно сотрудничать с патологами, чтобы убедиться, что очаги поражения изучены на предмет наличия злокачественной трансформации (2).
Дерматологи и медсестры дерматологических отделений должны периодически наблюдать пациентов, для контроля рецидивов (2). Междисциплинарный подход к наблюдению обеспечит наилучшие результаты лечения пациентов.
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии» Департамента здравоохранения Москвы, Москва, Россия
Консультативная поликлиника ГКБ №14 им. В.Г.Короленко, Москва
ГБУЗ "Московский научно-практический центр дерматовенерологии и косметологии" Департамента здравоохранения Москвы
ГУ МОНИКИ им. М.Ф. Владимирского
Порокератоз и его клинические варианты
Представлены собственные наблюдения нескольких клинических вариантов порокератоза, описаны генетические с аутосомно-доминантным типом наследования и средовые факторы, включающие интенсивность инсоляции, сопутствующие заболевания, лекарственно индуцированную иммуносупрессию, способствующие его возникновению. Согласно современной классификации, выделяют локализованные и диссеминированные формы. К локализованным относят порокератоз Мибелли, линейный порокератоз, ладонно-подошвенный точечный, генитальный, перианальный порокератоз; к диссеминированным — поверхностный актинический, поверхностный диссеминированный, диссеминированный ладонно-подошвенный, систематизированный линейный порокератоз. Многообразие форм порокератоза при идентичности гистологических признаков позволяет считать их клиническими вариантами одного заболевания. Методы лечения включают как наружную терапию — цитостатические препараты, аналоги витамина D, иммуномодуляторы, ингибиторы кальциневрина, ретиноиды, так и системную — ретиноиды, а также хирургическое иссечение очагов, криотерапию, электродиссекцию и кюретаж образований, дермабразию очагов и фотодинамическую терапию. Кроме того, пациенты должны быть информированы об использовании солнцезащитных средств и эмолиентов. Выбор метода лечения определяется формой заболевания, локализацией, прогнозом, а также косметическими показаниями. Важны своевременная диагностика и наблюдение за пациентами, страдающими порокератозом (особенно линейной формой), и лицами с иммуносупрессией, поскольку риск возникновения плоскоклеточного рака кожи в очагах у таких пациентов повышен.
Порокератозы — гетерогенная группа редких хронических заболеваний с преимущественным поражением эпидермиса по типу гиперкератоза.
Этиология и патогенез. Дерматоз наследуется аутосомно-доминантно с неполной пенетрантностью гена, семейные случаи с той или иной частотой наблюдаются при всех клинических вариантах порокератоза. Впервые связь между развитием диссеминированного актинического порокератоза и локусом 12-й хромосомы была установлена в 2000 г. при исследовании семейного случая заболевания в Китае [1]. В настоящее время имеются данные о наличии нескольких локусов хромосом (12q23.2—24.1, 12q21.2—24.21, 18p11.3, 1p31.3—p31.1и 16q24.1—24.3, 12q24.1—24.2, 15q25.1—26.1), ответственных за развитие диссеминированного поверхностного актинического порокератоза и диссеминированного порокератоза ладоней и подошв [2—5]. При ограниченных формах порокератозов, таких как порокератоз Мибелли и зостериформный порокератоз, имеет место мозаицизм, при котором соматические мутации приводят к локальной потере гетерозиготности [6]. Группа исследователей из Китая [7] обнаружили явное подтверждение связи между мутацией в гене мевалонаткиназы (МVК) и диссеминированным поверхностным актиническим порокератозом. Мевалонаткиназа — это фермент, кодируемый геном MVK. Данный фермент участвует в мевалонатном метаболическом пути, который принципиально важен для синтеза множества биологически активных соединений, необходимых для клеточного метаболизма. При исследовании влияния экспрессии на активность кератиноцитов установлено, что MVK-ген участвует в кальций-индуцируемой дифференциации кератиноцитов и MVK-экспрессия способна защитить данные клетки от апоптоза, инициируемого в результате действия ультрафиолетовых лучей спектра А. Кроме того, MVK играет важную роль в дифференцировке кератиноцитов, регулируя экспрессию кератина-1 в шиповатом слое эпидермиса и инволюкрина в гранулярном слое.
Солнечная инсоляция, PUVA-терапия, лучевая терапия являются триггерными факторами развития диссименированного актинического порокератоза и порокератоза Мибелли [8]. Иммуносупрессия, ассоциированная с порокератозом, может быть связана с приемом различных лекарственных препаратов: преднизолона и азатиоприна, этанерцепта и адалимумаба, иммуномодулирующих препаратов у реципиентов органных трансплантантов [9—13]. Частота развития порокератоза у пациентов после трансплантации органов составляла 1—11% [14]. Описаны случаи развития порокератоза у пациентов с ВИЧ-инфекцией, сахарным диабетом, заболеваниями печени и злокачественными новообразованиями различных органов [15—20]. Связь между порокератозом и иммуносупрессией до конца не ясна. Возможно, иммуносупрессия может стимулировать экспрессию мутантного клона клеток как напрямую, так и опосредованно через процесс эпидермальной дифференцировки [21]. Эпидермальные клетки Лангерганса в очагах на коже реципиентов почечного трансплантанта снижают выработку антигенов HLA-DR, что вызывает нарушение местного иммунитета, обусловливающее порокератоз [22].
Клинические проявления
По клинической картине заболевания выделяют пять основных форм порокератоза:
1. Классический вариант порокератоза Мибелли.
2. Линейный (зостериформный) порокератоз.
3. Поверхностный диссеминированный актинический порокератоз.
4. Поверхностный диссеминированный эруптивный порокератоз.
5. Диссеминированный ладонно-подошвенный порокератоз.
Некоторые авторы [23, 24] выделяют также гигантский порокератоз, кератотический веррукозный порокератоз, эруптивный зудящий порокератоз. Однако точечный ладонно-подошвенный порокератоз Манту относят к ладонно-подошвенным кератодермиям, а не к кератозам.
Классический вариант порокератоза Мибелли чаще наблюдают у представителей мужского пола. Дерматоз может возникать в любом возрасте: от первых месяцев жизни до старости. Преимущественная локализация — тыл кистей и стоп (рис. 1),
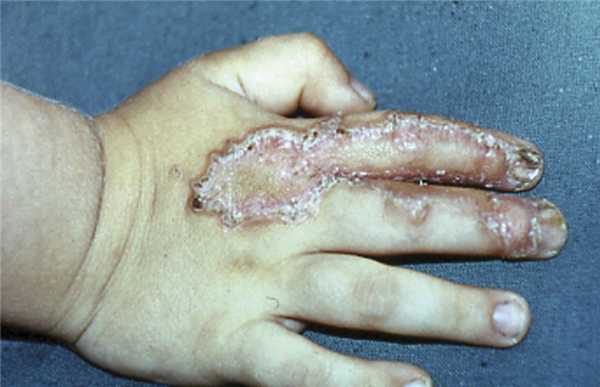
Рис. 1. Классический вариант порокератоза Мибелли. но встречается и на других участках, включая кожу туловища, слизистые оболочки полости рта и половых органов. Расположение очага на волосистой части головы завершается рубцовой алопецией. Начальными проявлениями обычно бывают мелкие (до 0,5 см в диаметре) безболезненные узелки конической формы серовато-коричневого оттенка. В центре элемента видно углубление с роговой пробкой, а края приподнимаются в виде кольцевидного рогового валика. Постепенно увеличиваясь в размерах, элемент за несколько лет превращается в бляшку, обычно не превышающую в диаметре 5 см, но иногда и значительно больше, до 15×25 см [25]. Края очагов четкие, образованные роговым валиком коричневого цвета характерной конфигурации: внутренняя часть поднимается вертикально, наружная — более пологая. На отдельных участках валика виден желобок. Толщина краев значительно варьирует. На тыле стопы, подвергающейся трению обувью, или на гигантских бляшках ширина валика может достигать 1 см и выглядеть настоящим рогом. На закрытых участках (кожа туловища) край два виден и лучше определяется при пальпации. Бордюр может быть фрагментированным, прерывистым. Центральная часть иногда не отличается от нормальной кожи. Чаще (особенно на бляшках большого размера) она атрофичная, блестящая, лишенная волос. Кожа в очаге пигментируется или покрывается телеангиэктазиями. Иногда центр становится сквамозным, с выраженным кератозом. Классическая форма порокератоза Мибелли может проявляться как солитарной бляшкой, так и отдельными сгруппированными более мелкими элементами, разделенными видимо неизмененной кожей. Их центр остается кератотическим или веррукозным. Субъективные ощущения обычно отсутствуют, потоотделение в этом участке снижено или отсутствует.
Линейный порокератоз отличается от классического зостериформным расположением кольцевидных бляшек по линиям Захарьина—Геда (рис. 2).
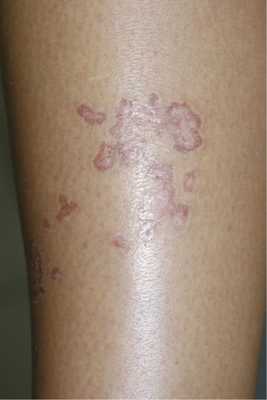
Рис. 2. Линейный (зостериформный) порокератоз. Количество элементов и протяженность такого поражения значительно варьируют, но все морфологические особенности порокератоза Мибелли сохраняются: запавшая центральная часть элементов и приподнятая периферическая зона в виде рогового валика, на котором заметна продольная полоска.
Поверхностный диссеминированный эруптивный порокератоз (вариант Респиги) характеризуется появлением множественных высыпаний, расположенных на различных участках кожного покрова. Чаще поражается кожа верхних и нижних конечностей, лица, затылка (рис. 3).
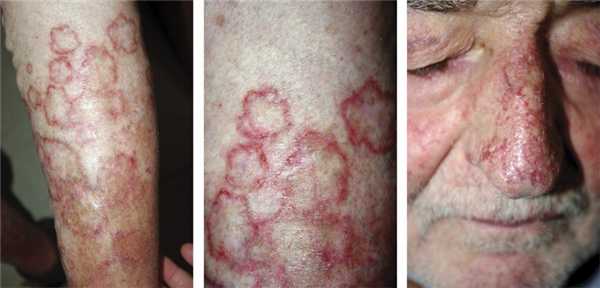
Рис. 3. Диссеминированный поверхностный эруптивный порокератоз. Элементы имеют четкие контуры, полициклические, неправильные или овальные очертания, диаметр от 2 до 4 см. Оси, проведенные через их длинный размер, располагаются, как правило, в определенном порядке: вертикально или горизонтально. Цвет желто-коричневатый, центр гладкий, атрофичный, слегка западающий по отношению к уровню окружающей кожи. Бордюр очень тонкий, блестящий в проходящем свете. От актинического порокератоза отличается более крупными размерами элементов, отсутствием строгой привязанности к открытым участкам тела и нередкими жалобами на зуд.
Поверхностный диссеминированный актинический порокератоз встречается значительно чаще классической формы Мибелли. Он возникает у предрасположенных субъектов обоих полов, но чаще у женщин, в возрасте от 30 до 60 лет, нередко диагностируется у нескольких членов одной семьи [26, 27]. Проявляется многочисленными мелкими, не превышающими в диаметре 5—8 мм пятнистыми элементами желтовато-коричневого цвета, с атрофичным центром, окруженным приподнятым гиперкератотическим узким валиком с бороздкой на поверхности (рис. 4).
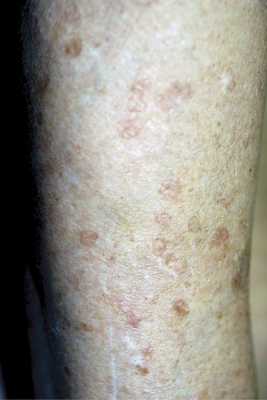
Рис. 4. Диссеминированный поверхностный актинический порокератоз. Иногда он настолько узок, что его присутствие легче определить при пальпации, а не визуально. Локализуется симметрично на открытых участках кожного покрова — голенях, предплечьях, кистях, реже на лице и шее. Ладони и подошвы никогда не поражаются. Клиническая своеобразность этого варианта порокератоза заключается в диссеминированном характере высыпаний, их мелких размерах, отсутствии склонности к слиянию и образованию бляшек, локализации на открытых солнечным лучам участках кожных покровов. В отличие от классической формы порокератоза, чаще регистрируемой у представителей мужского пола, актинический вариант преобладает у женщин. Патогенетическая роль солнечного облучения подтверждается случаями обострения процесса в летнее время: периферический бордюр утолщается, центр розовеет и пигментируется, иногда возникает зуд, появляются новые элементы. В конце зимы отмечается склонность к побледнению элементов, часть из которых становится едва заметной.
Ладонно-подошвенный диссеминированный порокератоз описан лишь в 1971 г. у 8 членов одной фамилии в четырех поколениях [28]. Элементы клинически и гистологически не отличаются от классической формы порокератоза Мибелли, но локализуются преимущественно на ладонях и подошвах. Некоторые бляшки с приподнятым бордюром могут достигать размеров нескольких сантиметров.
Патогистология
Диагностически значимые морфологические изменения при всех клинических вариантах порокератоза идентичны и расположены, как правило, в периферической части очага. Наиболее характерным признаком является так называемый cornoidlamella — особый тип ороговения, представленный столбикообразными структурами («колоннами») из компактных роговых масс с сохраненными ядрами, исходящими из инвагинации эпидермиса, эпидермальной части протока потовой железы или эпителия устья волосяного фолликула (рис. 5, 6).
Рис. 5. Cornoidlamella, исходящая из эпидермиса (а), протока потовой железы (б), эпителия устья волосяного фолликула (в). Окраска гематоксилином и эозином. ×200. Рис. 6. Лихеноидная воспалительная реакция при порокератозе. Эпидермис с вакуолизацией цитоплазмы клеток базального слоя и атрофией. В верхних отделах дермы — скопления меланофагов и коллоидных телец, лимфогистиоцитарная инфильтрация. Окраска гематоксилином и эозином. ×400. Под «колоннами» отсутствует зернистый слой, иногда отмечают вакуолизацию кератиноцитов шиповатого и базального слоев, дискератоз, дисплазию (от легкой до карциномы in situ). На других участках эпидермис может быть нормальной толщины, с акантозом и явлениями атрофии [29—31]. Воспалительная реакция при порокератозе может быть неспецифической и лихеноидного типа, гистологически напоминая атрофический красный плоский лишай или красную волчанку. Лихеноидная воспалительная реакция чаще встречается при актинической диссеминированной форме порокератоза. В верхних отделах дермы при этом могут наблюдаться меланофаги и коллоидные тельца [29, 30] (см. рис. 6). При иммуногистохимическом исследовании при порокератозе, как и при многих злокачественных опухолях, отмечают гиперэкспрессию ядрами кератиноцитов, расположенных под cornoidlamella, p53 белка-супрессора роста опухолей. Это может служить потенциальным объяснением онкогенеза при порокератозе, однако, в отличие от опухолей мутации гена p53, при порокератозе в настоящее время не выявлены [31, 32].
Диагноз и дифференциальный диагноз. Многообразие клинических проявлений порокератоза может затруднять своевременную диагностику. Начальные проявления способны симулировать бородавки, кожный рог, веррукозные невусы. Единичная бляшка классической формы порокератоза Мибелли обычно не представляет диагностических затруднений. Поверхностные формы имеют значительное сходство с атрофическим красным плоским лишаем: округлые очертания, пигментация и атрофия в центре, тонкий периферический ободок с легким блеском. В пользу порокератоза свидетельствуют отсутствие или слабая выраженность зуда, очень медленная эволюция элементов и характерная гистологическая картина.
Линейное расположение элементов порокератоза похоже на линейный веррукозный невус или линейное расположение красного плоского лишая.
Атрофические и пигментированные кольцевидные элементы на лице могут иметь значительное сходство с дискоидной красной волчанкой. Такие же элементы диаметром до 1,5 см с атрофическим центром и периферическим валиком наблюдают при так называемом субтропическом красном плоском лишае, нередко встречающемся у жителей Среднего Востока (Узбекистан, Туркмения, Египет, Ирак). Высыпания обычно возникают на открытых участках и рецидивируют в летнее время (рис. 7).
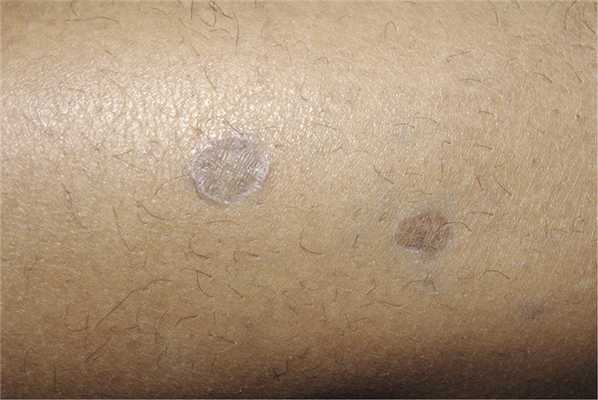
Рис. 7. Субтропический красный плоский лишай.
Актинический кератоз проявляется ограниченными сухими очагами гиперкератоза диаметром не более 1 см с сероватыми корками на поверхности. Типичная локализация — кожа лица (спинка носа, лоб), теменные области, лишенные волос, у мужчин (рис. 8).
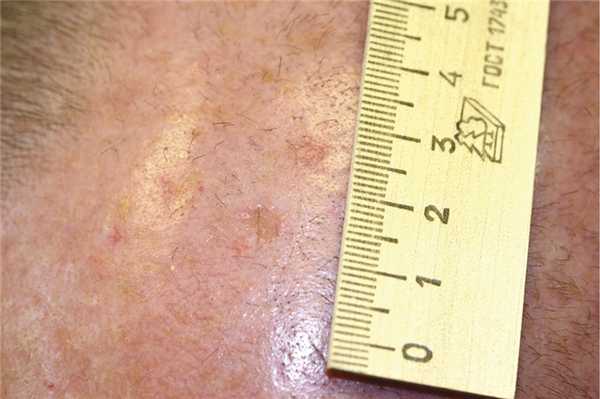
Рис. 8. Актинический кератоз. Встречается чаще у лиц со светлым типом кожи, подвергавшихся избыточной инсоляции. Возможна медленная трансформация в плоскоклеточный рак. Некоторые клинические формы порокератоза требуют дифференциального диагноза с ладонно-подошвенным порокератозом Манту и серпигинирующим кератозом Лутца—Мишера.
Порокератоз может проявляться также на слизистой оболочке полости рта: небе, языке, щеках, где клиническая картина напоминает красный плоский лишай, а также на слизистой оболочке половых органов и на губах. Сочетание таких высыпаний с кожными проявлениями облегчает диагностику, а изолированные случаи являются казуистически редкими.
Таким образом, многообразие форм порокератоза при идентичности гистологических признаков позволяет считать их клиническими вариантами одного заболевания, а не отдельными нозологическими формами, о чем сообщалось и ранее [33]. Это подтверждается также случаями сочетания у одного больного разных клинических вариантов порокератоза [29].
Эволюция элементов при классической форме очень медленная: они существуют десятилетиями, постепенно увеличиваясь в своих размерах за счет центробежного роста. Иногда у подростков или взрослых появляются новые высыпания. Это явление более характерно для лиц мужского пола, так как у женщин с возрастом наблюдаются случаи улучшения и даже исчезновения некоторых элементов во время беременности, а на их месте остается легкая атрофия. В случаях поверхностного диссеминированного или актинического порокератоза динамика клинических проявлений может быть значительно быстрее, в том числе и сезонная (улучшение в зимнее время). Иногда существенное улучшение наблюдают даже при кратковременном нанесении топических кортикостероидов [34]. В очагах порокератоза описаны случаи малигнизации с развитием плоскоклеточного рака, болезни Бовена и базально-клеточного рака кожи [35—38]. Больные всеми формами порокератоза, особенно с выраженным процессом, должны оставаться под наблюдением дерматолога из-за возможного злокачественного перерождения.
Лечение
Рекомендованные некоторыми авторами системные ретиноиды в дозе до 1 мг/кг/сут оказывают временный эффект, но при этом обладают значительным побочным действием. Из косметических соображений возможно разрешение некоторых элементов с помощью лазерного испарения, электрокоагуляции, криотерапии. При диссеминированном актиническом порокератозе необходимо применение фотозащитных средств.
Сведения об авторах
Ладонно-подошвенные кератодермии: причины, симптомы, диагностика, лечение
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Ладонно-подошвенные кератодермии - большая группа заболеваний, очень разных по своей морфологии. Некоторые из них являются самостоятельным заболеванием, другие входят в состав многочисленных синдромов, третьи представляют собой одно из проявлений диффузных кератозов. Гистогенетически все многообразие клинических проявлений можно свести к нескольким гисгоморфологическим типам.
Все ладонно-подошвенные кератодермии имеют общие гистологические признаки: в разной степени выраженный акантоз. гиперкератоз, иногда очаговый паракератоз; изменения в базальном слое эпидермиса и базальной мембране отсутствуют. Воспалительной реакции в дерме, как правило, нет, лишь иногда встречаются небольшие периваскулярные инфильтраты в верхней ее части. К особенностям, позволяющим разделять ладонно-полошвенные кератодермии на различные типы, относятся изменения структуры зернистого и шиповатого слоев эпидермиса: гиперкератоз с увеличением количества слоев зернистого слоя (гранулез), эпидермолитический гиперкератоз. атрофия или отсутствие зернистого слоя. Гиперкератоз и гранулез отмечаются при подавляющем большинстве ладонно-подошвенных кератодермий, как при диффузных, так и при ограниченных формах.
К диффузным формам кератодермии относят следующие нозологические единицы.
Ладонно-подошвенная кератодермия Тоста-Унны
Наследуется по аутосомно-доминангному типу, характеризуется диффузным поражением ладоней и подошв. Описаны также изменения в области межфаланговых суставов кистей. Существует с рождения или развивается в течение 1-го года жизни, редко - в более позднем возрасте Имеется диффузный кератоз ладоней и подошв с полоской застойной зритемы но его краю. Часты болезненные трещины.
Патоморфология. Выраженный гиперкератоз, гранулез, гипертрофия потовых желез, иногда картина эпидермолитического гиперкератоза, но в таких случаях необходимо исключать ограниченную форму буллезной ихтиозиформной эритродермии. При электронно-микроскопическом исследовании выявлены атипичные кератогиалиновые гранулы двух типов - менее электронно-плотные гранулярного строения и более электронно-плотные, прикрепленные к первым.
Ладонно-подошвенная кератодермия Вернера
Наследуется по аутосомно-доминантному типу. Выявлена мутапия гена, кодирующего кератин 9, расположенного в локусе 17ql2-q21. Заболевание развивается в первые недели жизни. Клиническая картина сходна с ладонно-подошвенной кератодермией Тоста-Уины. Отмечаются гипергидроз и утолщение ногтевых пластинок. Описана самопроизвольная отслойка роговых масс, происходящая 1-2 раза в год.
Патоморфология. Сходна с таковой при врожденной буллезной ихтиозиформной эритродермии,. что подтверждено методом электронной микроскопии. Можно предполагать, что в основе гистогенеза заболевания лежит нарушения образовании тонофибрилл. Биохимический анализ выявил в эпидермисе появление низкомолекулярных кератинов, свидетельствующих о нарушении дифференцировки эпителиоцитов.
Мутилируюшая кератодермия
Наследуется по аутосомно-доминантному типу, характеризуется наличием кератоза с сотовидной поверхностью на ладонях и подошвах, кератотических очагов звездчатых очертаний на тыле кистей кистей и стоп, внутренней поверхности лучезапястных суставов, кольцевидных перетяжек пальцев (псевлоайнгум). Часто встречаются ониходистрофии, описана диффузная алопепия.
Сотовидный кератоз, но без перетяжек, наблюдается также при ладонно-подошвенной кератодермии, ассоциированной с глухотой, при которой, как и при мутилирующей ладонно-подошвенной кератодермии, на тыле кистей и стоп имеются кератотические очаги с переходом на внутреннюю поверхность лучезапястных суставов.
Патоморфология: гиперкератоз с гипергранулезом.
Диффузная ладонно-подошвенная кератодермия
С аутосомно-доминантным типом наследования (генный докус - 17q23-ater) может сочетаться с раком пищевода (синдром Howel-Evans), Кератоз развивается обычно в 5-15 лет, рак пищевода - после 30 лет. Одновременно могут наблюдаться множественные базалиомы.
Кератодермия острова Меледа
Син. болезнь острова Мелела наследуется по аутосомно-репессивному типу. Клинически характеризуется диффузным кератозом ладоней и подошв, выраженной воспалительной реакцией в виде эритематозного венчика вокруг кератотических очагов с выходом поражения на тыльную поверхность кистей и стоп, области коленных и локтевых суставов, нижней трети предплечий и голеней (в виде "перчаток и носков"). Часты контрактуры и сращения пальцев. Описано сочетание с псевдоайнгумом. Заболевание сопровождается гипергидрозом и изменениями ногтевых пластинок, возможны и лейкокератозы.
Патоморфология. При электронной микроскопии выявляют кератогиалиновые гранулы сложного строения, состоящие из менее плотной гранулярной сердцевины и более плотной периферической зоны, связанной с тонофиламентами. Подобные гранулы чаще располагаются в эпителиоцитах, расположенных в области устьев потовых желез.
Близка по клиническим проявлениям к болезни острова Меледа кератодермия, описанная A. Greither (1952). Однако эта форма наследуется по аутосомно-доминантному типу, отличается менее выраженным гиперкератозом, наличием на других участках кожного покрова изменений, сходных с наблюдаемыми при эритрокератодермиях, менее тяжелым течением, улучшением с возрастом.
Кератодермия Папийона-Лефевра
Син. синдром Папийона-Лефевра наследуется по аутосомно-рецессивному типу. Клиническая картина сходна с кератодермией острова Меледа. Поражение кожи сочетается с пародонтозом, воспалением десен и сосочков языка и подверженностью различным инфекционным заболеваниям. Иногда наблюдаются отставание в росте, гипотрихоз, кальцификация мозговых оболочек, сочетание с врожденными бронхоэктазами.
Патоморфология: массивный компактный гиперкератоз и гипергранулез; в эритематозно-сквамозных очагах в области крупных суставов и тыльной поверхности кистей и стоп гистологическая картина напоминает красный отрубевидный волосяной лишай (болезнь Девержи): гиперкератоз с чередованием участков орто- и паракератоза, неравномерный акантоз, незначительная периваскулярная воспалительная инфильтрация в сосочковом слое дермы.
Синдром Олмстеда
Представляет собой сочетание диффузной ладонно-подошвенной кератодермии с четкими краями, ониходистрофии, констрикции пальцев и периорифициального кератоза. В дополнение к перечисленным признакам описаны универсальная алопеция, лейкокератоз, аномалии зубов.
Ограниченные ладонно-подошвенные кератозы
Собирательный термин, использующийся для всех ограниченных (очаговых, линейных) форм кератодермии. Тип наследования аутосомно-доминантный. Клинические проявления заболевания могут появиться в юношеском возрасте или у взрослых. При крупноочаговых формах кератодермии на ладонях и подошвах обнаруживают монетовидные округлые кератотические очаги, наиболее выраженные на местах давления, и крупные, изолированные или сочетающиеся с линейными кератозами очаги в области сгибательных поверхностей пальцев. Может наблюдаться сочетание со спиралевидными курчавыми волосами. При электронно-микроскопическом исследовании в одном случае обнаружены отечность эпителиоцитов, повышение плотности тонофиламентов в супрабазальной области, вакуолизация шиповатых клеток, изменения в строении кератогиалиновых гранул и липидные капли в роговом слое.
Папулезные ладонно-подошвенные кератодермии отличаются рассеянным характером и меньшими размерами кератотических очагов. Развиваются в первые годы жизни (кератодермия Брауэра) или в 15-30-летнем возрасте (кератодермия Бушке-Фишера). Клинически характеризуются множественными плоскими, полушаровидными или веррукозными очажками ороговения, округлых или овальных очертаний, располагающимися обычно изолированно по всей поверхности ладоней и подошв, а не только в местах давления. После удаления роговых масс остается кратеро- или блюдцеобразное углубление. A. Greither (1978) считает перечисленные формы папулезных кератодермии идентичными.
Точечная врожденная акрокератодермия
Син. точечный кератоз ладоней и подошв характеризуется появлением на ладонях и тыле кистей мелких кератотических папул цвета нормальной кожи с гладкой блестящей поверхностью. Гистологически F.C. Brown (1971) выявил паракератотические столбики, сходные с наблюдаемыми при порокератозе Мибелли. D.G. Robestria и соавт. (1980) обнаружили при электронной микроскопии внутриядерные нарушения в виде множественных гипертрофированных ядрышек в клетках базального и шиповатого слоев, которые, по мнению авторов, способствуют развитию гиперкератоза. Описано сочетание этого заболевания с раком внутренних органов. M.J. Costello и R.C. Gibbs (1967) рассматривают папулезные и точечные кератодермии как синонимы.
Кератодермия с просвечивающими папулами представляет собой, возможно, разновидность точечной врожденной акрокератодермии. Наследуется также по аутосомно-доминантному типу, характеризуется желтовато-белыми, просвечивающими папулами с гладкой поверхностью, иногда с точечными углублениями в центре, сливающимися в бляшки. Сочетается с тонкими волосами на голове и атопией.
Точечный кератоз ладонных линий характеризуется наличием на ладонях и подошвах мелких гиперкератотических пробок, располагающихся в углублениях кожных линий, болезненных при надавливании.
Ладонно-подошвенная кератодермия с перекрученными волосами - аутосомно-доминантно наследуемое заболевание, характеризующееся наличием на ладонях и подошвах округлых очагов кератоза. Патологические изменения волос подтверждены методом сканирующей электронной микроскопии. В волосах гистохимически обнаружен дефицит цистеина.
Синдром Рнхнера-Ханхарта
Син.: кожно-глазной тирозиноз, тирозинемия II типа характеризуется болезненными ладонно-подошвенными кератотическими очагами, герпетиформной дистрофией роговицы и умственной отсталостью. Без лечения с возрастом развивается диффузная кератодермия, могут быть пузыри. Тип наследования аутосомно-рецессивный, поражен генный локус 16q22.1-q22. Гистологически, помимо общих для этой группы кератодермии признаков, выявляют эозинофильные включения в клетках шиповатого слоя. При электронно-микроскопическом исследовании находят увеличение количества тонофиламентов в шиповатых эпителиоцитах, тубулярные каналы а пучках тонофиламентов. В основе гистогенеза лежит дефицит фермента тирозина аминотрансферазы, что приводит к накоплению тирозина в крови и тканях. Предполагается, что молекулы L-тирознна мот способствовать образованию дополнительных поперечных связей. Это приводит к сгущению тонофибрилл в эпителиоцитах.
Ладонно-подошвешшя нумулярная кератодермия
Так называемые болезненные омозолелости наследуется по аутосомно-доминантному типу. Развивается в детском или молодом возрасте, характеризуется наличием ограниченных крупных гиперкератотических очагов. локализующихся в местах давления: на подошвах. у основания и на боковых поверхностях пальцев ног, на кончиках пальцев рук, болезненных при надавливании. Описаны пузыри по краям очагов, подногтевой или околоногтевой гиперкератоз, утолщение ногтевых пластинок и очаги гиперкератоза на голенях. Гистологически наблюдается эпидермолитический гиперкератоз.
Акрокератоэластоилоз Коста
Развивается в детском возрасте. Клинически проявляется мелкими, иногда сливающимися папулами птотноватой консистенции, сероватого цвета, просвечивающими, с блестящей поверхностью, располагающимися на ладонях и подошвах, по краям пальцев, в области пяточного сухожилия. Гистохимически в дерме в очагах поражения выявляются утолщение и фрагментация пластических волокон, электронно-микроскопически - изменения аморфной части их, нарушение расположения микрофибрилл. Изменения в зернистом слое отсутствуют.
Следует отметить, что большая группа ладонно-подошвенных кератодермяй до сих пор не классифицирован ни клинически, ни гистологически. В литературе имеются морфологические описания только отдельных случаев. В связи с этим диагностика, особенно дифференциальная, указанных заболеваний представляет большие трудности.
Различия в клинической характеристике высыпаний и типе наследования, особенности течения заболеваний внутри выделенных нами групп позволяют предположить неодинаковый их патогенез при сходной гистологической картине.
Читайте также: