Причины и классификация альбинизма у ребенка
Добавил пользователь Morpheus Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Альбинизм: причины появления, симптомы, диагностика и способы лечения.
Определение
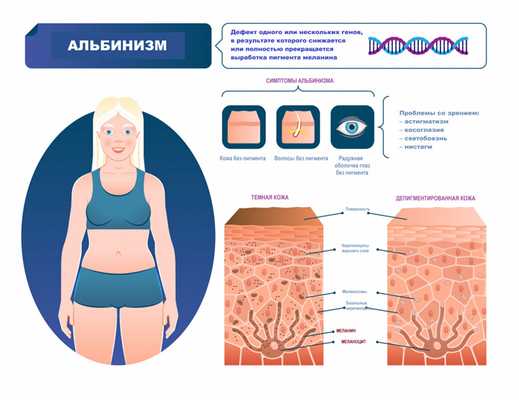
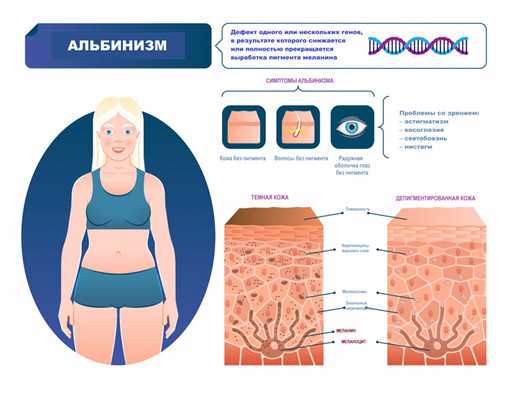
Альбинизм (от лат. albus — белый) рассматривается как наследственное заболевание и характеризуется врожденным частичным или полным отсутствием в организме пигмента меланина. Таким образом, кожа, волосы и радужная оболочка глаз выглядят обесцвеченными, бледными или имеют более светлый оттенок, чем должны были быть в норме.

Аномалия встречается примерно у 1 из 10-20 тысяч человек на земном шаре и не зависит от этнической принадлежности и пола.
Причины появления альбинизма
Цвет кожи детерминирован генетически. Наличие альбинизма говорит о мутации генов (на настоящий момент описаны 19 генов, отвечающих за появление альбинизма), ответственных за цвет кожи, волос и радужной оболочки глаз. Эти мутации приводят к недостаточной активности фермента тирозиназы, благодаря которой синтезируется аминокислота тирозин, являющаяся основным регулятором производства пигмента меланин - так схематично и очень упрощенно выглядит меланогенез.
Заболевание передается по аутосомно-рецессивному (то есть оба родителя должны являться носителями рецессивного гена) и Х-сцепленному (только в случае с геном GRP143) типам наследования.
Классификация заболевания
Ранее случаи альбинизма разделяли только по внешним (фенотипическим) проявлениям - полный и неполный.
К первому относились все типы альбинизма с выраженными нарушениями пигментации глаз, кожи и ее придатков. К неполным формам - глазные типы заболевания, когда отмечался недостаток пигментации радужной оболочки, а также те разновидности патологии, которые приводили к пятнистости кожи.
В настоящее время принята классификация, основанная на генетических аспектах:
Глазокожный альбинизм тип 1А - полное отсутствие пигмента меланина в организме.
Глазокожный альбинизм тип 1В - при этом типе синтезируется дефектный фермент тирозиназа, поэтому синтез меланина возможен, но с имеет разную степень выраженности.
Температурно-чувствительный глазокожный альбинизм - активность синтеза пигмента зависит от температуры тела. Для него характерно падение активности фермента тирозиназы, если температура тела более 37̊С. При этом типе альбинизма преобладают поражения глаз, поскольку их температура всегда выше 37̊С.
Глазокожный альбинизм тип 2 - активность фермента тирозиназы, нормальная, но нарушен транспорт тирозина, в результате чего меланин не синтезируется.
Глазокожный альбинизм тип 3 - встречается только у африканцев, формируется коричневая окраска кожного покрова.
Глазной альбинизм рецессивный и связанный с Х-хромосомой - синтез пигмента частично нарушен.
Аутосомно-рецессивный глазной альбинизм - данный тип не удалось связать с какими-либо генетическими нарушениями.
Кроме изолированных глазного и глазокожного типов существуют редкие наследственные синдромы альбинизма, которые связаны с системными поражениями. К ним относятся синдром Германски-Пудлака и синдром Чедиака-Хигаси, встречающиеся, к счастью, крайне редко.
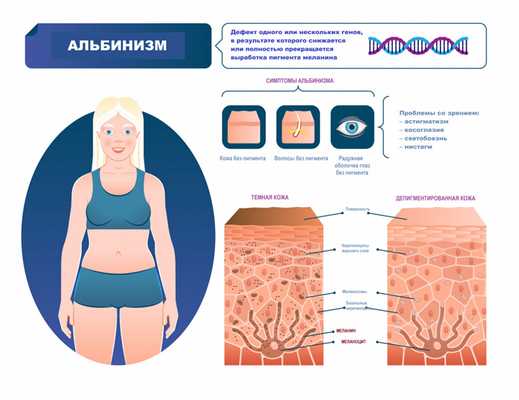
Симптомы альбинизма
Признаки альбинизма, как правило, очевидны, однако могут быть выражены слабо (это зависит от типа альбинизма и дефектного гена). При видимых нарушениях обесцвечены и кожа, и волосы, и радужная оболочка глаз. Один из важных симптомов альбинизма - зрительные нарушения:
- нистагм - быстрые, непроизвольные возвратно-поступательные движения глаз;
- косоглазие - неспособность обоих глаз оставаться направленными в одну и ту же точку или двигаться в унисон;
- экстремальная близорукость или дальнозоркость;
- светобоязнь - чувствительность к свету, иногда переходящая в дневную слепоту;
- астигматизм - аномальная кривизна передней поверхности глаза или внутренней части глаза, вызывающая снижение зрительной функции.
Диагноз ставится сразу после рождения ребенка. Врач-педиатр оценивает состояние пигментации кожи и волос, а офтальмолог - наличие той или иной зрительной патологии.
Подтвердить диагноз альбинизма может врач-генетик, проведя секвенирование генов. Очень важно выявить наследственный характер патологии и собрать анамнез.
Редким и дорогостоящим методом, но позволяющим уточнить прогноз заболевания, является определение тирозиназной активности в тканях (например, в волосяных фолликулах).
Чем лучше сохранена активность тирозиназы в тканях, тем ниже выраженность других симптомов альбинизма.
К каким врачам обращаться
Диагностикой альбинизма занимается врач дерматолог, но первым обнаруживает признаки альбинизма педиатр . При наличии патологии зрения необходима консультация офтальмолога.
Лечение альбинизма
На сегодняшний день специфического лечения альбинизма не существует, поэтому усилия врачей направлены на коррекцию остроты зрения больных и профилактику злокачественных патологий кожи.
- Из-за отсутствия меланина, выполняющего защитную функцию, люди с альбинизмом имеют высокий риск рака кожи.
- Со стороны органов зрения: снижение остроты зрения, отслойка сетчатки.
Специфической профилактики не разработано, есть только профилактические мероприятия, позволяющие улучшить качество жизни больного. Пациентам с альбинизмом рекомендовано защищать глаза от солнечного света солнцезащитными очками. Не рекомендуется находиться на ярком солнце и в любом случае следует защищать кожу специальными солнцезащитными средствами с высоким SPF.
Важно регулярно проходить осмотры дерматолога и офтальмолога для снижения риска развития осложнений.
- Кадышев В.В., Ряжская СА., Халанская О.В. и др. Клинико-генетические аспекты альбинизма // Клиническая офтальмология. - 2021;21(3). - С. 175-180. DOI: 10.32364/2311-7729-2021-21-3-175-180
- Бутов Ю. С. Дерматовенерология. Национальное руководство. Краткое издание / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова. - М.: ГЭОТАР-Медиа, 2020. - 896 с.
- Лоскутов И. А. Симптомы и синдромы в офтальмологии / Лоскутов И. А., Беликова Е. И., Корнеева А. В. - М.: ГЭОТАР-Медиа, 2021. - 256 с.
Альбинизм
Буллезный эпидермолиз
Буллезный эпидермолиз - группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10

Общие сведения
Буллезный эпидермолиз - это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса - десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии - пограничного буллезного эпидермолиза - являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз - имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз - делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз - имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи - эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе - киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно - развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты - механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств - типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность - как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Альбинизм - группа наследственных патологий, характеризующихся нарушениями или полным отсутствием пигментации кожи, волос, радужной оболочки глаза. Основными симптомами заболевания является очень светлая кожа и волосы, голубой или красноватый цвет глаз, в ряде случаев могут быть нарушения зрения. Диагностика альбинизма производится на основании настоящего статуса пациента, а также генетических исследований. Специфического лечения альбинизма на сегодняшний день нет, используют паллиативную терапию (коррекция зрения), а также существует ряд рекомендаций больным, как вести себя на солнце, защищать кожу и снизить вероятность осложнений.

Альбинизм - совокупность генетических патологий, при которых нарушаются процессы формирования или накопления пигмента меланина в клетках кожи, ее придатков, радужной оболочке и сетчатке глаза. Это состояние известно с древних временен, поражает лиц любой национальности или расы. Однако частота встречаемости альбинизма отличается у разных народностей - она составляет от 1:10000 до 1:2000000. Также этот показатель неодинаков для разных форм заболевания, которые в последние годы классифицируются по генетическим признакам. Ранее выделяли всего две формы альбинизма (глазную и кожно-глазную), хотя современная генетика различает, по меньшей мере, семь различных типов патологии. Кроме того, существует ряд наследственных заболеваний, симптомокомплекс которых, помимо всего прочего, включает в себя и альбинизм - например, синдром Чедиака-Хигаси, болезнь Германски-Пудлака.
Причины альбинизма
Основная причина развития альбинизма - нарушения метаболизма аминокислоты тирозина, и, как следствие, полный блок или ослабление синтеза и отложения пигмента меланина. К этому состоянию могут привести различные мутации генов, которые прямо или косвенно участвуют в процессе образования меланина. Например, наиболее тяжелая форма альбинизма - глазокожная 1А - обусловлена сложной мутацией гена alb-OCA1, расположенного на 11-й хромосоме. Он кодирует последовательность фермента тирозиназы, и при nonsense-мутациях его производство в организме полностью останавливается. В результате образование меланина также полностью прекращается, что и становится причиной тяжелого глазокожного альбинизма. Наследуется это состояние по аутосомно-рецессивному механизму.
Другой тип этого заболевания - глазокожный альбинизм 1В - обусловлен нарушением того же гена alb-OCA1, однако при этом его функционирование продолжается. С данной патологией ассоциировано более 50-ти мутаций вышеуказанного гена, каждая из которых в разной мере влияет на активность тирозиназы. Поэтому выраженность симптомов при глазокожном альбинизме 1В также очень вариабельна - от почти полного отсутствия меланина в тканях до слегка более светлого оттенка кожи и волос. В ряде случаев больные с такой формой патологии способны загорать, у них с возрастом темнеют волосы, могут появляться пигментные невусы. Интересным подтипом данного заболевания является температурно-чувствительный альбинизм, при котором активность тирозиназы резко падает при температуре свыше 37 градусов. Это приводит к тому, что пигментация сильнее проявляться на более холодных участках тела - кисти, стопы. Тогда как голова, глаза, подмышечные впадины часто остаются практически без пигмента.
Глазокожный альбинизм типа 2 является наиболее распространенной разновидностью данной патологии. Однако при этом генетические нарушения не затрагивают синтез тирозиназы, который сохраняется на достаточном уровне, активность и структура фермента также не страдают. Альбинизм такого типа обусловлен мутацией гена, расположенного на 15-й хромосоме. Предположительно, он кодирует протеин (Р-белок) мембраны меланосом, который отвечает за транспорт тирозина. При этой форме альбинизма проявления дефицита меланина также очень вариабельны, к тому же, пигментация может усиливаться со временем. Причины такого явления до сих пор не выяснены. Наследуется глазокожный альбинизм типа 2 по аутосомно-рецессивному типу.
Другая форма заболевания, глазокожный альбинизм типа 3 встречается почти исключительно у негроидной расы. При нем генетическими исследованиями выявлены мутации гена TRP-1, расположенного на 9-й хромосоме. Аналогичный ген у мышей отвечает за коричневую окраску шерсти, его функции у человека достоверно неизвестны. Предполагается, что он контролирует образование черной фракции меланина (эумеланина), а нарушение его структуры ведет к преимущественному синтезу коричневой разновидности пигмента. Как и другие глазокожные формы альбинизма, тип 3 передается по аутосомно-рецессивному механизму.
Каждый тип альбинизма характеризуются не только исчезновением меланина из кожи и ее придатков, но и зрительного аппарата глаза - радужной оболочки и пигментного слоя. Это приводит к нарушениям рефракции и прозрачности роговицы, астигматизму и косоглазию, фовеолярной гипоплазии сетчатки. Имеются формы альбинизма (так называемые глазные типы), которые характеризуются только поражением органов зрения. Наиболее распространенная форма глазного альбинизма передается по рецессивному типу и сцеплена с Х-хромосомой. Она обусловлена мутацией гена GPR143, кодирующего рецептор к G-белку меланоцитов глаз. В результате этого нарушаются процессы формирования меланосом, что и становится причиной развития глазного альбинизма. В 1970-м году была также выявлена аутосомно-рецессивная форма этого заболевания, однако патогенез данного типа до сих пор не определен - часть (14%) таких больных имели мутации гена alb-OCA1, другие (36%) - нарушения в гене Р-белка. Почти у половины пациентов с аутосомно-рецессивным глазным альбинизмом выявить генетическую причину заболевания не удалось.
Классификация альбинизма
Ранее все случаи альбинизма разделяли только по фенотипическим проявлениям - полный и неполный. К первому относились все типы глазокожного альбинизма, которые характеризуются выраженными нарушениями пигментации глаз, кожи и ее придатков. К неполным формам относили глазные типы заболевания, а также те разновидности патологии, которые приводили к пятнистости кожи. В настоящее время чаще используют генетическую классификацию, в рамках которой выделяют такие виды альбинизма:
- Глазокожный альбинизм тип 1А - его причиной выступает nonsense-мутация гена alb-OCA1, которая попросту «выключает» его экспрессию. В результате этого полностью останавливается синтез тирозиназы в организме.
- Глазокожный альбинизм тип 1В - так же, как и в предыдущем случае, он обусловлен мутациями гена alb-OCA1, однако при этом его экспрессия возможна. В результате синтезируется дефектный фермент тирозиназа с разной степенью активности. Выраженность проявлений такого альбинизма зависит от типа мутации гена.
- Температурно-чувствительный глазокожный альбинизм - является разновидностью типа 1В, характеризуется изменчивой активностью тирозиназы, которая зависит от температуры. Кожные проявления умеренные, тогда как офтальмологические нарушения могут быть значительно выражены. Данные особенности такого альбинизма обусловлены более высокой температурой глаз - следовательно, тирозиназа в них менее активна.
- Глазокожный альбинизм тип 2 - вызван мутацией гена, кодирующего Р-белок, являющийся элементом мембраны внутриклеточных меланосом. В результате транспорт тирозина в клетке нарушается, и синтез меланина не происходит даже при нормальной активности тирозиназы.
- Глазокожный альбинизм тип 3 - является следствием мутаций гена TRP-1, который, предположительно, контролирует образование эумеланина. Встречается только у африканцев, вызывает развитие коричневой окраски кожи и волос и умеренные офтальмологические нарушения.
- Глазной альбинизм рецессивный и связанный с Х-хромосомой. Он обусловлен мутацией гена GPR143, отвечающего за некоторые элементы внутриклеточной передачи информации.
- Аутосомно-рецессивный глазной альбинизм - его пока не удалось связать с конкретными генетическими нарушениями. Предполагается, что часть случаев такого заболевания является глазными формами глазокожных типов патологии - 1В и 2.
Даже в пределах одного генотипа альбинизма возможны значительные различия в степени выраженности симптомов. Это связано с тем, что мутации разного типа неодинаково влияют на выработку меланина.
Симптомы альбинизма
К главным проявлениям альбинизма относят бледность кожных покровов, особенно заметную при рождении больного ребенка. Нередко кожа имеет розоватый оттенок из-за просвечивающихся кровеносных сосудов, глаза при рождении голубые, но в некоторых ракурсах также могут иметь красноватый цвет. В дальнейшем, в процессе роста, симптомы альбинизма могут несколько изменяться в зависимости от типа заболевания. При типе 1А, который является наиболее тяжелым, синтеза меланина в организме не происходит вообще, поэтому у больного пожизненно сохраняется белый цвет кожи и волос и голубые глаза. Альбинизм типа 1В характеризуется быстрым накоплением в волосах желтого пигмента, поэтому они принимают светло-соломенный цвет, часто с возрастом происходит пигментация ресниц и роговицы глаз.
Температурно-чувствительный альбинизм часто проявляется своеобразным распределением меланина - нормальная пигментация наблюдается на конечностях, тогда как кожа головы остается бледной, волосы также сохраняют белый цвет. Глаза по причине повышенной, нежели на конечностях, температуры остаются у таких больных голубого цвета. Значительной переменчивостью симптомов характеризуется и альбинизм типа 2 - от почти полного отсутствия пигментации до незаметного посветления кожи и волос. Также для такой формы заболевания часто характерно улучшение синтеза меланина с возрастом - начинают темнеть волосы, появляться веснушки, возникать загар. Однако с пребыванием на солнечном свете необходимо быть осторожным - кожа больных альбинизмом крайне чувствительна к ультрафиолетовому облучению, легко возникают ожоги кожи и фотодерматиты.
Характерным симптомом альбинизма является нарушение остроты зрения у больных и другие офтальмологические изменения. Снижение зрения тем выраженнее, чем слабее синтезируется в организме меланин, особенно в роговице и пигментном слое сетчатки. Кроме этого, частыми спутниками альбинизма являются косоглазие, астигматизм, нистагм, которые возникают сразу при рождении или в первые годы жизни. При глазных формах заболевания подобные симптомы проявляются без нарушения пигментации кожи и волос. Из-за отсутствия защитного слоя меланоцитов у больных альбинизмом часто возникает светобоязнь, иногда переходящая в дневную слепоту.
Диагностика
Определение альбинизма во многих случаях возможно сразу же после рождения больного - дерматолог, оценивая состояние пигментации кожи и волос способен выявить заболевание и приблизительно узнать его разновидность. Дальнейшее наблюдение у этого специалиста необходимо для мониторинга течения патологии и для профилактики возможных осложнений - например, рака кожи. Врач-офтальмолог при альбинизме нередко выявляет прозрачность радужной оболочки, у взрослых больных часто определяется гипоплазия сетчатки в области желтого пятна. Фовеолярный рефлекс резко снижен или отсутствует. У лиц с неполным альбинизмом на глазном дне часто обнаруживаются очаги депигментации. Обнаруживаются и другие нарушения зрения - нистагм, астигматизм, миопия.
Для подтверждения диагноза и уточнения типа патологии врач-генетик может назначить секвенирование ассоциированных с ней генов. Также важную роль играет составление наследственного анамнеза, возможна генетическая диагностика родственников больного для выявления носителей дефектных генов. Редким и дорогостоящим методом диагностики альбинизма является определение тирозиназной активности в тканях (например, в волосяных фолликулах), но это позволяет несколько уточнить прогноз заболевания. Чем лучше сохранена активность этого пигмента в тканях, тем ниже выраженность других симптомов этой патологии.
Дифференциальную диагностику альбинизма следует проводить с другими наследственными патологиями, которые сопровождаются сходными кожными и офтальмологическими симптомами. В первую очередь это синдромы Чедиака-Хигаси и Германского-Пудлака, Х-сцепленный ихтиоз, микрофтальмия, болезнь Каллмана.
Лечение и прогноз альбинизма
Специфического лечения альбинизма на сегодняшний день не существует, разработаны лишь профилактические мероприятия, позволяющие улучшить качество жизни больного. Для сохранения существующего уровня зрения необходима защита глаз от солнечного света - это достигается ношением специальных солнцезащитных очков или контактных линз. Появления на ярком солнце необходимо избегать или же защищать кожу специальными кремами и лосьонами. Если придерживаться этих рекомендаций, то в целом прогноз альбинизма благоприятный - больные могут прожить долгую и полноценную жизнь. При этом необходимы регулярные консультации дерматолога и офтальмолога - для профилактики осложнений, таких как рак кожи или отслойка сетчатки.
Читайте также: