Примеры семейной эксудативной витриоретинопатии симулирующей ретинобластому
Добавил пользователь Morpheus Обновлено: 22.01.2026
Ретинопатия недоношенных (РН) является основной причиной слепоты у детей, рождающихся в цивилизованных странах. Это пролиферативное заболевание, причиной которого является незрелость структуры сетчатки у преждевременно рождённых детей. Неонатология развивается семимильными шагами и теперь даже у глубоко недоношенных новорождённых сохраняются высокие шансы на выживание. Вместе с этим растёт количество тяжёлых форм ретинопатии. Патогенез развития заболевания до конца не изучен, несмотря на то, что в этой области проводилось много исследований.
В развитии ретинопатии недоношенных особую роль играют 2 периода жизни ребёнка:
- 1 фаза наступает, если ребёнок родился и начал дышать между 22-й и 30-й неделей постконцептуального возраста, неразвитая сетчатка становится относительно гипероксической, а ретинальные сосуды прекращают расти.
- 2 фаза начинается между 31-34-й неделей постконцептуального возраста из-за огромного количества факторов роста и окислительных процессов в клетках, которые провоцируют неуправляемый рост сосудов сетчатки.
Сначала между сосудами и бессосудистой тканью клетчатки появляется линия, а за ней гребень ткани. Затем патологические сосуды прорастают в гребень и в полость стекловидного тела, прогрессирование состояния приводит к образованию рубцов, что и является причиной тракционного отслоения сетчатки.
Обычно при исследовании ретинопатии недоношенных врачи-офтальмологи не берут во внимание доклиническую стадию, а рассматривают только рубцовую или активную. Отечественные исследователи Сидоренко Е.И. и Николаева Г.В. в 2011 году устранили этот пробел, установив период и патогенез, когда развивается ретинопатия недоношенных.
Учёные обнаружили 5 факторов, которые влияют на развитие первой доклинической стадии ретинопатии недоношенных, а именно:
- избыток кислорода в сосудистой части сетчатки разрушает кислородозависимый гипоксический фактор (HIF-1) ангиогенеза;
- стекловидное тело накапливает излишек кислорода;
- интенсивная кислородная помощь (подключение к ИВЛ, дополнительная оксигенация, пребывание в кувезе);
- незрелая ауторегуляция капилляров в сосудистой части сетчатки, которая при контакте с кислородом, становится причиной тяжелого ангиоспазма;
- сопутствующая патология недоношенного ребенка.
В работе отмечается, что чем раньше малыш появляется на свет, тем чаще у него бывают спазмы сосудов. Также исследователи пришли к выводу, что созревание ауторегуляции сосудов сетчатки относится примерно к 30 неделям постконцептуального возраста. Авторы нашли решение ещё одной проблемы: если своевременно лечить сопутствующие заболевания у новорождённых, это поможет снизить тяжесть ретинопатии недоношенных.
Основной проблемой в лечении РН до сих пор считается отсутствие новых методов лечения, несмотря на то, что регулярно открываются новые возможности. Учёные считают, что у недоношенных детей образуются кислородсодержащие свободные радикалы, которые активируют эндотелий сетчатки и приводят к развитию ретинопатии недоношенных. Моноклональные и нейтрализующие антитела в процессе проведения клинических испытаний показали себя, как средства, борющиеся с воспалениями.
Генетические испытания, проводившиеся на мышах, подтвердили, что малая GTPase RhoB, вырабатывающаяся от стресса, является предпосылкой для патологического ангиогенеза. Исследователи считают, что различные механизмы, контролирующие активность GTPase, обеспечивают большие возможности для накапливания лекарства в определённой области.
Учёные из США и Китая в процессе эксперимента выявили, что ангиогенный фактор плейотропин активизируется в стекловидном теле больных с пролиферативной ретинопатией. Демонстрация проводилась на мышах. В результате выявлено, что интравитреальная инъекция антител положительно влияет на состояние сетчатки, а именно, предотвращает ретинопатию, сосудистые разрастания и неоваскуляризацию сетчатки.
Антитела, нейтрализующие плейотропин, повлияли на хориоидальную неоваскуляризацию в сторону улучшения (эксперимент с мышами), из чего можно сделать вывод, что плейотропин ухудшает сосудистое состояние хориоидеи. Вследствие эксперимента доказан проангиогенный эффект плейотропина (Ptn) на состояние сосудов и его участие в развитии глазных болезней у мышей. Учёные утверждают, что плейотропин можно использовать в качестве мишени для лечения антителами (Ab).
Учёные из Индии долгое время работали над изучением генетического фактора развития ретинопатии недоношенных. Они совершили прорыв в науке, благодаря картированию генов и изучению частот аллелей и генотипов, неравновесные сцепления и оценки гаплотипов, и доказали, что гены CFH, CFB, FBLN5, CETP и CXCR4 связаны с развитием ретинопатии недоношенных.
Эти данные были получены в процессе исследования недоношенных детей с диагнозом РН и без него. Проводился количественный анализ цитокинов и белков-кандидатов, полученных из стекловидного тела, а также слёз у детей с тяжелой формой ретинопатии недоношенных.
Для этой цели применялись следующие виды оценивания:
- вестерн-блоттинг,
- мультиплексные массивы гранул,
- зимография.
В процессе исследования выявлено, что факторы, вызывающие воспаление (C3, C4, IL-1ra и другие) не просто влияют на развитие ретинопатии недоношенных, но оказывают воздействие на разные стадии развития этого заболевания. Исследователи пришли к выводу, что высокий уровень активного комплемента в стекловидном теле, вызывает микроглию, из-за чего усиливается воспалительный процесс. Изучение патогенеза заболевания помогает проанализировать и подсчитать количество маркеров воспаления в слёзной жидкости, что позволяет составить ранний прогноз развития ретинопатии новорождённых и своевременно назначить терапию, облегчающую состояние пациента.
Японские учёные проводили эксперимент, где изучали генетические аномалии, которые являются причиной развития и тяжести ретинопатии недоношенных. Поскольку клиническая картина семейной экссудативной витреоретинопатии (FEVR) и ретинопатии недоношенных очень похожа, учёные набрали группу больных, страдающих тяжелой ретинопатией недоношенных, и проверили их на генетические мутации 3-х генов, которые отвечают за сигнальный путь рецептора wingless/int1 (Wnt) - FZD4.
Таким образом, 17 больных из контрольной группы прошли обследование методами ПЦР и прямого секвенирования генов ND, FZD4 и LRP5. Результаты эксперимента оказались интересными: у одного больного определяется мутация в гене LRP5, у второго больного определяется мутация в гене ND.
Учёные предположительно утверждают, что генетические аномалии в сигнальной системе рецептора Wnt в процессе роста сетчатки, могут являться причиной развития тяжелой формы ретинопатии недоношенных.
Эксперимент также наводит на мысль, что тяжелая ретинопатия недоношенных может быть полигенетической. Если сравнить генетические нарушения между тяжелыми и легкими случаями ретинопатии недоношенных, то мы, возможно, поймём этиологию развития этого заболевания. Исследователи высказывают предположение, что нужно и дальше исследовать эту область, чтобы понять, относятся ли эти генетические дефекты впервые выявленным в роду или являются наследственными. Так или иначе, генетические исследования позволяют на ранних стадиях выявлять случаи высокого риска и давать прогноз тяжести течения заболевания.
Последние эксперименты в изучении патогенеза ретинопатии недоношенных привели офтальмологов к возможности идентифицировать некий временной период и механизмы, которые являются спусковым механизмом развития ретинопатии недоношенных. Поэтому теперь многие специалисты уделяют внимание именно доклинической стадии развития заболевания. Они уже вплотную приблизились к возможности раннего выявления и профилактики РН.
Выявляя сопутствующие заболевания у младенцев, которые могут повлиять на патогенетический путь развития ретинопатии новорождённых и, изучая механизмы их действия, учёные пришли к тому, что эффективность скрининга значительно увеличилась. В скором будущем появится возможность генетического консультирования по профилактике развития ретинопатии недоношенных, что поможет выявлять потенциальные группы риска.
Подобные исследования дают надежду на решение проблем, связанных с лечением и профилактикой ретинопатии недоношенных, но требуют дальнейшего изучения патогенеза этого заболевания.
© Виджияпала К.Ш., Николаева Г.В., 2022. Полная версия статьи в журнале «Российская детская офтальмология», № 1 2022.
Экссудативная семейная витреоретинопатия

Экссудативная семейная витреоретинопатия является наследственным заболеванием, которое передается по аутосомно-доминантному типу. При этом отсутствует перфузия в периферических областях сетчатки, в результате чего возникает неоваскуляризация.
Этиология заболевания
Обычно у детей с семейной экссудативной витреоретинопатией не имеется других аномалий развития. Эти новорожденные доношенные, не имеют респираторных нарушений, то есть и не выполнялась гипероксигенация.
Клиника заболевания может различаться и, несмотря на двустороннее поражение, часто отмечается асимметричность симптомов. При выраженных отклонениях возникает косоглазие или белый зрачковый рефлекс, характерные для младенческого возраста. Снижение зрения может проявляться в любом возрасте.
Отличительные симптомы
Классическим симптомом экссудативной семейной витреоретинопатии является отсутствие кровотока в периферических ретинальных капиллярах. На границе между васкуляризированной областью сетчатки и периферической зоной формирется неоваскуляризация. Дополнительно из-за сокращения фиброваскулрной ткани происходит стягивание сетчатки. Иногда можно обнаружить гемофтальм, возникший при кровоизлиянии в вещество стекловидного тела.
В ряде случаев формируется экссудативная и регматогенная отслойка сетчатки, субретинальная или интраретинальная липидная экссудация. Все это нередко носит асимметричный характер.
Сопутствующие проявления
При тяжелом течении семейной экссудативной витреоретинопатии вознкают:
- Катаракта;
- Отслойка сетчатки;
неоваскулярная глаукома; - Лентовидная кератопатия;
- Атрофия глазного яблока.
Иногда эти проявления сочетаются у одного пациента.
Дифференциальная диагностика
Если у пациента обнаружена отслойка сетчатки, то следует учитывать и другие причины, помимо семейной экссудативной витреоретинопатии, которые могли бы вызвать соответствующие изменения в младенческом возрасте.
При офтальмоскопии обычно удается выявить типичные изменения глазного дна. Одновременно требуется обследование всех членов семьи, так как при бессимптомном течении также выявляются аномалии в сетчатке.
Прогноз и лечение
Если заболевание проявилось в раннем возрасте, то более характерны тяжелые изменения сетчатки. Чтобы предотвратить прогрессирование неоваскуляризации, можно провести фотокоагуляцию или криотерапию периферической сетчатки, не содержащей капилляров.
В сложных случаях, когда возникла отслойка сетчатки глаза, выполняют склеральное пломбирование или витрэктомию. Важно также обследовать всех членов семьи, так как прогрессирование заболевания может начаться в любом возрасте.
Экссудативная витреохореоретинальная дистрофия
Exudative vitreoretinopathy, familial, X-linked recessive (EVR2, FEVR; MIM 305390) - Х-сцепленное рецессивное заболевание, характеризующееся тракцией сетчатки, периферическим помутнением стекловидного тела и субретинальными и интраретинальными экссудатами. Тракционное действие на сетчатку оказывают мембраны, находящиеся за светопреломляющими средами и прикрепляющиеся к реснитчатому телу соединительнотканными тяжами. В основном семейная экссудативная ретинопатия наследуется по аутосомно-доминантному типу. Однако имеются семьи с Х-сцепленным рецессивным типом наследования.
При EVR2 обнаружены мутации в гене NDP (Norrie Disease Protein; MIM 300658), в котором находят мутации, приводящие к болезни Норри (MIM310600). Ген локализован на хромосоме Хp11.4 и состоит из трех экзонов. Кодирующими являются экзоны 2 и 3. Обнаружено пять мутаций в гене NDP, приводящие к EVR2. Однако есть доказательства существования по крайней мере еще одного локуса для EVR2 на Х-хромосоме.
В Центре Молекулярной Генетики проводится поиск мутаций кодирующих экзонах гена NDP методом прямого автоматического секвенирования.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Клинические проявления семейной экссудативной витреоретинопатии у детей при нарушении нуклеотидной последовательности гена FZD4
Семейная экссудативная витреоретинопатия (СЭВР) — редкое генетически гетерогенное заболевание, имеющее разные типы наследования (аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный) и широко варьирующие клинические проявления. До 40 % случаев развития СЭВР связаны с мутациями гена FZD4.
Цель работы — анализ клинических проявлений СЭВР у детей при нарушении нуклеотидной последовательности гена FZD4.
Материал и методы. В НМИЦ ГБ им. Гельмгольца и МГНЦ им. академика Н.П. Бочкова совместно обследованы 18 пациентов в возрасте от 3 нед до 17 лет с диагнозом СЭВР. Углубленное офтальмологическое обследование включало детальную офтальмоскопию в условиях медикаментозного мидриаза, ультразвуковое и электрофизиологическое исследование, фотофиксацию изменений глазного дна с помощью RetCam и Fundus Foto. Молекулярно-генетическое обследование проведено методом прямого секвенирования по Сэнгеру.
Результаты. Нарушение нуклеотидной последовательности гена FZD4 обнаружено у 3 (16,7 %) пациентов из 2 неродственных семей. В одной семье у девочки 12 лет первые симптомы офтальмологической патологии (снижение зрения, косоглазие) выявлены в 3,5 года. Во второй семье манифестация клинической картины мутации гена FZD4 у 2 детей отмечена на первом году жизни (в возрасте 5 и 11 мес).
Заключение. Клиническая картина у 3 пациентов с выявленными изменениями нуклеотидной последовательности гена FZD4 характеризуется ранней манифестацией и двусторонним асимметричным офтальмоскопическим поражением. Полученные результаты указывают на необходимость тщательной своевременной диагностики СЭВР у детей раннего возраста, междисциплинарного подхода к изучению заболевания, что внесет свой вклад в понимание патогенеза, разработку диагностического и лечебно-реабилитационного алгоритма.
Ключевые слова
Об авторах
Людмила Анатольевна Катаргина — д-р мед. наук, профессор, начальник отдела патологии глаз у детей, заместитель директора по научной работе
ул. Садовая-Черногрязская, д. 14/19, Москва, 105062
Виталий Викторович Кадышев — канд. мед. наук, старший научный сотрудник лаборатории генетической эпидемиологии, заведующий кафедрой офтальмогенетики Института высшего и дополнительного профессионального образования
ул. Москворечье, д. 1, Москва, 115522
Екатерина Валерьевна Денисова — канд. мед. наук, врач-офтальмолог, старший научный сотрудник отдела патологии глаз у детей
Елизавета Александровна Гераськина — аспирант отдела патологии глаз у детей
Андрей Владимирович Марахонов — канд. биол. наук, старший научный сотрудник лаборатории генетической эпидемиологии, доцент кафедры медицинской генетики Института высшего и дополнительного профессионального образования
Софья Айдаровна Гарифуллина — младший научный сотрудник лаборатории генетической эпидемиологии
ФГБУ «НМИЦ глазных болезней им. Гельмгольца» Минздрава России; ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова»
Россия
Инна Владимировна Зольникова — д-р мед. наук, старший научный сотрудник отдела клинической физиологии зрения им. С.В. Кравкова, профессор кафедры офтальмогенетики Института высшего и дополнительного профессионального образования
ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова»; ФГБНУ «Национальный НИИ общественного здоровья им. Н.А. Семашко»
Россия
Рена Абульфазовна Зинченко — д-р мед. наук, профессор, заведующая лабораторией генетической эпидемиологии, заместитель директора по научно-клинической работе, главный научный сотрудник отдела исследований общественного здравоохранения
ул. Воронцово Поле, д. 12, стр. 1, Москва
Список литературы
1. Criswick V.G., Schepens C L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 1969; 68 (4): 578-94.
2. Pendergast S.D., Trese M.T. Familial exudative vitreoretinopathy: results ofsurgical management. Ophthalmology. 1998; 105: 1015-23.
4. Gitter K.A., Rothschild H., Waltman D.D., Scott B., Azar P. Dominantly inherited peripheralretinal neovascularization. Arch. Ophthalmol. 1978 Sep; 96 (9): 1601-5. doi: 10.1001/archopht.1978.03910060235009
5. Li Y., FuhrmannC., SchwingerE.,Gal A.,LaquaH.The gene for autosomal dominant exudative vitreoretinopathy (Criswick-Schepens) on the long arm of chromosome 11. Am J Ophthalmol. 1992; 113 (6): 712-3. doi: 10.1016/s0002-9394(14)74800-7
6. Toomes C., Bottomley H. M., Scott S., et al. Spectrum and frequency of FZD4 mutations in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2004; 45 (7): 2083-90. doi: 10.1167/iovs.03-1044
7. Ells A., Guernsey D. L., Wallace K., et al. Severe retinopathy of prematurity associated with FZD4 mutations. Ophthalmic Genetics. 2010; 31 (1): 37-43. doi: 10.3109/13816810903479834
8. Kondo H., OhnoK.,TahiraT., et al. Delineation of the critical interval forthe familial exudative vitreoretinopathy gene by linkage and haplotype analysis. Hum. Genet. 2001; 108 (5): 368-75. doi: 10.1007/s004390100503
9. Muller B., Orth U., Van Nouhuys C.E., et al. Mapping of the autosomal dominant exudative vitreoretinopathy locus (EVR1) by multipoint linkage analysis in four families. Genomics. 1994; 20 (2): 317-9. doi: 10.1006/geno.1994.1176
10. Chen C., Sun L., Li S., et al. The spectrum of genetic mutations in patients with asymptomatic mild familial exudative vitreoretinopathy. Exp. Eye Res. 2020; 192: 107941. doi: 10.1016/j.exer.2020.107941
11. Wang Z., Chen C., Sun L., et al. Symmetry of foldsin FEVR:Agenotype-phenotype correlation study. Exp. Eye Res. 2019; 186: 107720. doi: 10.1016/j.exer.2019.107720
12. Han S., Sun J., Yang L., Qi M. Role of NDP- and FZD4-Related Novel Mutations Identified in Patients with FEVR in Norrin/ -Catenin Signaling Pathway. BioMed Research International. 2020; 2020: 7681926 doi: 10.1155/2020/7681926
13. Gilmour D.F. Familial exudative vitreoretinopathy and related retinopathies. Eye (Lond). 2015; 29 (1): 1-14. doi: 10.1038/eye.2014.70
14. Salvo J., Lyubasyuk V., Xu M., et al. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients.Invest. Ophthalmol. Vis. Sci. 2015; 56 (3): 1937-46. doi: 10.1167/iovs.14-16065
15. Rao Feng-Qin, Cai Xue-Bi, Cheng Fei-Fei, et al. Mutations in LRP5 , FZD4 , TSPAN12 , NDP, ZNF408, or KIF11 genes account for 38.7% of Chinese patients with familial exudative vitreoretinopathy. Invest. Opthalmol. Vis.Sci. 2017. 58 (5): 2623-9. 10.1167/iovs.16-21324
16. HiroyukiK, Eiichi U., Shunji K., Koichiro H. Risk allele of the FZD4 gene forfamilial exudative vitreoretinopathy. Ophthalmic Genetics. 2018; 39 (3): 405-6. doi: 10.1080/13816810.2017.1401090
17. Wang X., Feng Y., Li J., et al. Retinal diseases caused by mutations in genes not specifically associated with the clinical diagnosis. PLoS One. 2016; 11 (10): e0165405. doi: 10.1371/journal.pone.0165405
18. Wang Y., Rattner A., Zhou Y., et al. Norrin/frizzled 4 signaling in retinal vascular development and blood brain barrier plasticity. Cell. 2012; 151 (6): 1332-44. doi: 10.1016/j.cell.2012.10.042
19. Xu Q., Wang Y., Dabdoub A., et al. Vascular development in the retina and inner ear: control by Norrin and frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004; 116 (6): 883-95. doi: 10.1016/s0092-8674(04)00216-8
20. Qin M., Hayashi H., Oshima K., et al. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathywith mutationsin the LRP5 and/ or FZD4 genes. Hum Mutat. 2005; 26 (2): 104-12. doi: 10.1002/humu.20191
21. Ranchod T.M., Ho L.Y., Drenser K.A., et al. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology. 2011; 118 (10): 2070-5. doi: 10.1016/j.ophtha.2011.06.020
22. Toomes C., Bottomley H. M., Scott S., et al. Spectrum and frequency of FZD4 mutations in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2004; 45 (7): 2083-90. doi: 10.1167/iovs.03-1044
23. Nallathambi J., Shukla D.,Rajendran A., et al.Identification of novel FZD4 mutations in Indian patients with familial exudative vitreoretinopathy. Mol. Vis. 2006; 12: 1086-92.
24. Niehrs C., Acebron S. P. Mitotic and mitogenic Wnt signalling. EMBO J. 2012; 31 (12): 2705-13. doi: 10.1038/emboj.2012.124
25. Nishimura M., Yamana T., Sugino M., et al. Falciform retinal fold as sign of familial exudative vitreoretinopathy. Jpn. J. Ophthalmol. 1983; 27 (1): 40-53. Jpn J Ophthalmol. 1983; 27 (1): 40-53.
26. van Nouhuys C.E. Dominant exudative vitreoretinopathy and other vascular developmental disorders of the peripheralretina. Doc. Ophthalmol. 1982; 54 (1-4): 1-414. doi: 10.1007/BF00183127
27. Nishina S., Suzuki Y., Yokoi T., et al. Clinical features of congenital retinal folds. American journal of ophthalmology. 2012; 153 (1): 81-7. doi: 10.1016/j.ajo.2011.06.002
Рецензия
При поддержке: Молекулярно-генетическое обследование выполнено при финансовой поддержке РНФ (проект № 17-15-01051) и в рамках государственного задания Министерства образования и науки РФ.
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Клинико-радиологические аспекты ретинобластомы
Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии Росздрава,
Российская детская клиническая больница, Москва, Россия.

УЗИ сканер HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Введение
Ретинобластома - наиболее частая внутриглазная опухоль у детей младшего возраста, это быстро растущая опухоль эмбрионального типа, вариант исходящей из незрелой сетчатки нейробластомы. Раннее адекватное лечение существенно сказывается на прогнозе [2, 3]. При строго интраокулярной локализации опухоли выздоровление наступает более чем в 90 % случаев [4]. Однако диагностика часто запаздывает, так как педиатры, наблюдающие детей младшего возраста, недостаточно знакомы с проблемой и с запозданием направляют детей на радиологическое обследование и в специализированные центры. В связи с этим мы представляем результаты собственных наблюдений и критический анализ литературы.
Материалы и методы
По итогам работы поликлинического отделения РДКБ за 1999-2006 гг. среди 7311 детей, доставленных на прием к окулисту, было 8 детей с ретинобластомой. Во всех наблюдениях дети были в возрасте до 3 лет, ретинобластома имелась спорадическая, односторонняя. Наряду с офтальмологическим обследованием проводили радиологические диагностические вмешательства.
Результаты исследований
Ребенку с подозрением на ретинобластому после сбора анамнеза, общего клинического обследования, внешнего осмотра глаза, биомикроскопии с помощью щелевой лампы и непрямой офтальмоскопии необходимо выполнить дополнительные инструментальные исследования, прежде всего УЗИ, флюоресцентную ангиографию, рентгеновскую компьютерную томографию (РКТ) или магнитно-резонансную томографию (МРТ).
На первом месте по простоте использования и быстроте получения информации стоит УЗИ. Исследование позволяет достаточно быстро отдифференцировать ретинобластому от неопухолевых образований. Ретинобластома имеет характерные ультразвуковые признаки. При А-сканировании определяется высокоамплитудный эхосигнал с аттенуацией в нормальных тканях орбиты. По нашим данным, при В-сканировании с частотой излучения не менее 5-7 Мгц визуализируется округлое или неправильной формы внутриглазное образование с отложениями кальция. Образование исходит из задних отделов глаза, хорошо отграничено от стекловидного тела (рис. 1). Кровоток в опухоли не определяется. Эхографические исследования особенно показаны в динамике для оценки реакции опухоли на радиотерапию. Ультразвуковая картина опухоли весьма близка к так называемым псевдоопухолям сетчатки - гранулематозном изменении сетчатки, например, при токсокарозе. Безапелляционное заключение в совокупности с некритическим восприятием картины глазного дна по данным офтальмоскопии может привести к абсолютно не показанной энуклеации.
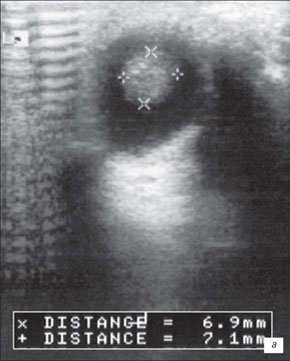
а) Небольшая ретинобластома. В ткани опухоли видны более плотные мелкие структуры (включения кальция).
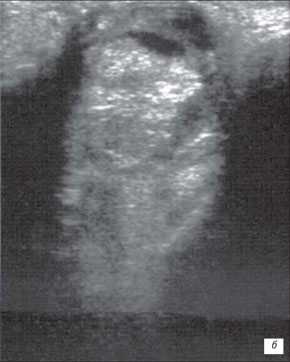
б) Большая ретинобластома. Опухоль заполняет практически всю заднюю камеру глаза. Структура опухоли неоднородна, средней эхогенности. Регистрируются множественные ярко белые (высокая эхогенность) отложения кальция..
Флюоресцентная ангиография особенно информативна при множественных внутриглазных опухолях. В случаях мелких внутриретинальных опухолей в артериальную фазу становятся видны незначительно расширенные немногочисленные питающие артерии, умеренная гиперваскуляризация в венозную фазу и небольшое свечение узла Р на завершающем этапе. В средних и больших узлах Р определяются расширенные питающие артерии и дренирующие вены, а сама опухоль оказывается окруженной большим количеством капилляров.
После ультразвуковых исследований ребенка направляют на КТ или МРТ, которые, как и эхография, выявляют очаги кальцификации в ткани опухоли, типичные, но не патогномоничные для ретинобластомы. Кальцинаты в сетчатке наблюдаются при ретинальном ангиоматозе, астроцитоме, инвазии нематодами и других заболеваниях. Преимуществом КТ по сравнению с УЗИ является способность метода выявить экстраокулярную экспансию опухоли и сопутствующую пинеалобластому (трилатеральную ретинобластому). Не менее важно, что КТ, как и МРТ, оказывается полезным для исключения псевдоопухолей, т.е. гранулематозных изменений сетчатки [5].
МРТ может оказаться принципиально важным вспомогательным методом диагностики ретинобластомы. В Т1-взвешенном изображении ретинобластома гиперэхогенна по отношению к стекловидному телу, хотя и с трудом отличима от него. В Т2-взвешенном изображении опухоль гипоэхогенна. В Т2 режиме лучше визуализируются и очаги кальцификации, экссудации и геморрагии [6]. По нашему мнению, у детей младшего возраста качество МРТ-изображения может уступать КТ по специфичности из-за меньшей вероятности выявления внутриопухолевых кальцинатов. МРТ высокоинформативна для диагностики экссудативной или геморрагической отслойки сетчатки. В Т1 и Т2 режимах это проявляется субретинальным сигналом, соответствующим по плотности стекловидному телу.
Известно, что ретинобластома может быть как наследственным заболеванием, так и спорадически возникшей опухолью. В 94 % наблюдений опухоль является спорадической, в 6 - семейной. Во всех случаях семейной Р пациенты по аутосомно-доминантному типу передают повышенный риск ее развития своим потомкам. В то же время не менее 5 % больных с односторонней спорадической ретинобластомой могут быть носителями гена ретинобластомы и передавать его своим потомкам [7]. У 25 % пациентов с семейной ретинобластомой обнаруживают интерстициальную делецию длинного плеча 13-й хромосомы (13q14) с потерей хромосомного материала. Утерянное хромосомное вещество включает в себя ген, кодирующий ядерный фосфопротеин, являющийся супрессором репликаций ДНК. В итоге теряется контроль клеточного цикла. Ретинобластома развивается у 80-90 % носителей данного хромосомного дефекта [1]. Для этого варианта характерны задержка умственного развития, низкий рост и дисморфии лица [8]. Считается, что у остальных пациентов с семейным вариантом ретинобластомы имеются мелкие, не выявляемые мутации в этом же регионе 13.й хромосомы. Пенетрантность гена - 80-100 %. Неполная пенетрантность - ложная, обусловленная случаями не диагностированной ретиноцитомы, доброкачественная опухоль.
В 75 % наблюдений опухоль односторонняя и средний возраст пациента на момент выявления опухоли - 12 месяцев. В 25 (по другим данным - в 50 %) наблюдений ретинобластома поражает оба глаза. При двухсторонней локализации риск передачи гена опухоли потомству велик. Средний возраст пациентов при двухсторонней локализации опухоли на момент ее манифестации составляет 23 месяца. Однако опухоль может обнаруживаться у новорожденных и крайне редко - у взрослых. Цитологически возможен полный спектр от низкодифференцированных до высокодифференцированных клеток. Для низкодифференцированной опухоли типичны мелкие или средних размеров круглые нейробласты с большим гиперхромным ядром и небольшим объемом цитоплазмы. Гистологически для случаев зрелой ретинобластомы характерно обнаружение розеток Флекснер.Винтерштайнера и флореток, напоминающих цветочные букеты (flores). Розетки Флекснер-Винтерштайнера составлены из цилиндрических клеток, кругообразно расположенных вокруг светлого центра. Розетки высокоспецифичны для ретинобластомы, однако встречаются и при других внутриглазных опухолях, особенно при медуллоэпителиоме. Флоретты выглядят как нежные эозинофильные структуры, состоящие из опухолевых клеток, содержащих эозинофильные отростки, которые вследствие проникновения через фенестрированную мембрану приобретают грушевидную форму (рис. 2). Кроме ретинобластомы флоретты обнаруживаются и в ретиноцитомах, доброкачественных вариантах ретинобластомы. Возможен частичный или полный кальциноз опухоли как исход ее некроза.
Читайте также: