Разработка лекарственного препарата
Добавил пользователь Евгений Кузнецов Обновлено: 02.02.2026
Многие применяемые в настоящий момент лекарственные препараты Обзор лекарственных препаратов По определению законодательства США лекарственные препараты (в отличие от продуктов питания или изделий медицинского назначения) — это любые вещества, предназначенные для диагностики, полного. Прочитайте дополнительные сведения были открыты в ходе экспериментов, которые проводились на животных и на людях. Однако в наше время многие лекарственные препараты разрабатываются с прицелом на конкретное заболевание. Сначала изучают аномальные изменения в организме на биохимическом и клеточном уровне, вызываемые заболеванием, что позволяет в дальнейшем разработать химические соединения, способные специфичным образом предотвратить или скорректировать такого рода аномальные изменения (путем взаимодействия с определенными функциональными участками организма — т.н. «сайтами»). Если новое соединение кажется перспективным, его структуру обычно многократно изменяют, чтобы:
В идеале лекарственный препарат является
Весьма сильным и эффективным: можно применять низкие дозы даже при заболеваниях, тяжело поддающихся лечению.
Эффективным при приеме внутрь (хорошо всасывается из пищеварительного тракта): простым в использовании.
Достаточно стабильным в тканях и жидкостях организма: таким образом, в идеале должно быть достаточно одной дозы в день (препараты краткосрочного действия могут быть предпочтительными при заболеваниях, которые требуют только краткосрочного лечения).
В ходе разработки исследуемого препарата определяются стандартные или средние дозы. Тем не менее, разные люди по-разному реагируют на лекарственные препараты. На терапевтический ответ оказывают влияние многие факторы, такие как возраст (см. Старение и лекарственные препараты Старение и лекарственные препараты Лекарственные препараты, представляющие собой наиболее распространенный способ медицинского вмешательства, являются важной частью медицинского обслуживания пожилых людей. Без лекарств многие. Прочитайте дополнительные сведенияЭтапы разработки лекарственного препарата
(Краткий обзор стадий разработки препарата представлен в таблице От лаборатории до аптеки От лаборатории до аптеки ).
Первоначальный этап разработки лекарственных препаратов
После того, как лекарственный препарат, могущий оказаться полезным в лечении определенных расстройств, был идентифицирован или разработан, его изучают на лабораторных животных (первоначальный этап разработки лекарственных препаратов). Первоначальный этап разработки лекарственных препаратов предусматривает сбор сведений о том, каким образом действует лекарственный препарат, насколько он эффективен и какие токсические эффекты он вызывает, в том числе возможные эффекты в отношении репродуктивной способности и здоровья потомства. На этой стадии многие лекарственные препараты отклоняются, поскольку они оказываются неэффективными или слишком токсичными.
Если на первоначальном этапе разработки лекарственный препарат оказывается перспективным, соответствующий экспертный совет организации (ЭСО) должен утвердить программу клинического исследования данного препарата, а в Управление США по контролю качества пищевых продуктов и лекарственных средств (FDA) подается заявка на регистрацию нового исследуемого препарата (IND). Если FDA утверждает данную заявку, лекарственный препарат можно испытывать на людях (данная фаза именуются «клинические исследования»).
Клинические исследования
Такого рода исследования состоят из нескольких фаз; к участию в них привлекаются исключительно добровольцы, давшие свое письменное согласие. Для одобрения FDA необходимы три фазы клинических исследований:
В ходе фазы 1 проводится оценка безопасности и токсичности препарата у человека. Небольшому количеству здоровых молодых испытуемых назначают препарат в различных дозах, чтобы определить, при какой дозе появляются первые признаки токсичности.
В ходе фазы 2 проводится оценка воздействия, которое оказывает данный препарат на то заболевание/расстройство, против которого он направлен, и определяется надлежащая его доза. Примерно 100 лицам, страдающим определенного рода заболеванием/расстройством, вводят различные дозы препарата, чтобы выяснить, дает ли это какую-либо пользу. Тот факт, что препарат эффективен у животных в ходе первоначального этапа разработки лекарственных препаратов, еще не означает, что он будет эффективен у человека.
В ходе фазы 3 проводятся испытания препарата на значительно большей (от сотен до тысяч людей) популяции лиц, страдающих определенного рода заболеванием/расстройством. Подбор этих участников производится таким образом, чтобы они были как можно более схожи с людьми, которые могут применять данный препарат в реальности. В ходе дальнейшего изучения эффективности препарата отмечаются новые присущие ему побочные эффекты. В ходе исследований фазы 3 обычно проводится сравнение нового лекарственного препарата с известным проверенным препаратом, с плацебо или же и с тем, и с другим.
Кроме определения эффективности лекарственного препарата, исследования на людях сосредоточены на видах и частоте возникновения побочных эффектов и на факторах, которые обуславливают склонность пациентов к возникновению такого рода эффектов (например, возраст, пол, наличие сопутствующих расстройств и применение других лекарственных препаратов).
Утверждение
Если в ходе исследования было установлено, что данный лекарственный препарат достаточно эффективен и безопасен, в FDA подается заявка на регистрацию нового лекарственного препарата (NDA) (включая данные исследований на животных и с участием людей, предполагаемый процесс производства лекарственного препарата, инструкция по медицинскому применению и этикетку препарата). После этого FDA изучает представленную информацию и принимает решение о том, достаточно ли препарат эффективен и безопасен, чтобы разрешить его реализацию на фармацевтическом рынке. После утверждения FDA данный лекарственный препарат станет доступным для применения. Этот процесс в общей сложности занимает до 10 лет. В среднем из 4000 лекарственных препаратов, которые изучаются в лабораториях, только около 5 изучаются на людях, и только 1 из этих 5 препаратов проходит утверждение и начинает применяться в медицинской практике.
В каждой стране имеется собственный процесс утверждения, который может отличаться от процесса, используемого в США. Если препарат одобрен для применения в одной стране, это совсем не означает, что он доступен для применения в другой стране.
Фаза 4 (пострегистрационный период)
После утверждения нового препарата иногда проводятся исследования фазы 4. Компания-производитель обязуется проводить наблюдение за применением препарата и немедленно уведомлять FDA о любых дополнительных, ранее не обнаруженных побочных эффектах. Всячески приветствуется участие в продолжающемся наблюдении за применением лекарственного препарата врачей и фармацевтов. Такого рода наблюдение имеет очень большое значение, поскольку до выхода препарата на рынок даже довольно обширные исследования позволяют выявить лишь относительно распространенные побочные эффекты (возникающие примерно у одного из 1000 человек). Важные побочные эффекты, возникающие у одного из каждых 10 000 (или более) человек препарата, можно обнаружить только в случае, когда данный препарат применяет большое число людей, т. е., после выхода препарата на рынок.
FDA может отозвать разрешение на применение препарата в случае появления новых данных о том, что данный препарат вызывает тяжелые побочные эффекты. Например, диетическое средство фенфлурамин было отозвано с рынка, поскольку у некоторых людей, принимавших его, возникли серьезные расстройства со стороны сердечно-сосудистой системы.
Разработка лекарственного препарата
Потенциальные лекарственные вещества могут быть найдены путем полномасштабного скрининга сотен и тысяч молекул на предмет наличия биологической активности. В других случаях знание специфических молекулярных аспектов патогенеза того или иного заболевания позволяет использовать рациональный подход к созданию новых препаратов путем компьютерного моделирования, либо модификации существующих фармакологически активных молекул.
В ходе ранних доклинических исследований потенциально активные соединения изучаются на животных для оценки желаемого действия и токсичности. Вещества, показавшие свою эффективность и безопасность, становятся кандидатами для дальнейшего изучения у человека. В США протокол, содержащий описание клинического исследования, должен быть утвержден Институциональным наблюдательным советом соответствующего учреждения и Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA), которое затем выдает разрешение на исследование нового лекарственного средства. С этого момента начинается период действия патента на препарат, обычно дающий собственнику эксклюзивные права на ближайшие 20 лет; однако лекарственное средство не может быть выпущено на рынок без одобрения FDA.
В ходе фазы 1 клинического исследования оценивается безопасность и токсичность препарата у человека. Для этого различные дозы изучаемой субстанции принимаются небольшим числом (обычно от 20 до 80) здоровых добровольцев (как правило, молодых мужчин) для определения дозы, при которой возникают первые признаки токсичности.
Целью фазы 2 является подтверждение активности препарата при конкретной патологии. Изучаемый препарат назначается для лечения либо профилактики данной патологии. Дополнительной задачей этой фазы является определение оптимального режима дозирования.
В исследованиях фазы 3 оценивается эффект препарата на более многочисленные (от 100 до нескольких тысяч человек) и гетерогенные группы пациентов для подтверждения возможности клинического использования изучаемого препарата. В ходе этой фазы также проводится сравнение препарата с существующими стандартными схемами терапии и/или плацебо. В исследование могут быть вовлечены практикующие врачи и многие лечебные учреждения. Основной целью этой фазы является подтверждение эффективности препарата и его возможных эффектов (как положительных, так и отрицательных), которые могут быть не выявлены в исследованиях 1-й и 2-й фаз.
Когда будут собраны достаточные данные для регистрации лекарственного средства, материалы подаются в контролирующую организацию, дающую разрешение на выпуск его на рынок. С момента ранней стадии разработки лекарственного средства до регистрации зачастую проходит около 10 лет.
Фаза 4 (постмаркетинговые наблюдения, фармаконадзор) наступает после того, как препарат был одобрен и поступил в продажу, и может включать в себя формальные научные исследования наряду с постоянно поступающими отчетами о побочных эффектах Побочные эффекты Само собой разумеется, что лекарственный препарат (как и любой курс лечения) должен назначаться исходя исключительно из его лечебного эффекта на пациента. Для этого необходимо учитывать способность. Прочитайте дополнительные сведения . Фаза 4 охватывает, как правило, более крупные популяции и более длительные периоды времени, чем фазы 1-3, что помогает выявить редкие или медленно проявляющиеся побочные эффекты, которые тяжело обнаружить в более коротких, малых исследованиях. Кроме того, в реальных условиях лекарства употребляют не только пациенты, которые удовлетворяют строгим критериям отбора, используемым в клинических испытаниях; препараты, как правило, принимают пациенты с высоким риском развития побочных эффектов. Часто в такие исследования включаются особые подгруппы пациентов (например, беременные, дети, пожилые пациенты). Некоторые препараты, одобренные FDA после фазы 3, были впоследствии сняты с продажи после выявления серьезных побочных эффектов в 4-й фазе исследования.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Путь лекарства

Обзор
Создание нового лекарства требует большого количества ресурсов и времени. Невозможно предугадать успешность результата: подходящее, казалось бы, вещество, может дать сбой на любом этапе.
Автор
Редакторы
- Драг-дизайн
- Здравоохранение
- Инфографика
- Медицина
- Наглядно о ненаглядном
- Фармакология
Инфографика на конкурс «био/мол/текст»: Казалось бы, для читателей «Биомолекулы» нет ничего понятнее, чем процесс создания лекарства. Однако почти никто не делал из этого инфографику — для смертных попроще. Вкратце — отсюда вы узнаете, сколько времени занимает процесс создания лекарства и насколько это недешево. И может быть, догадаетесь, что, если по телевизору сказали, что ученые обнаружили вещество, способное победить рак какую-нибудь заразу, то еще ох как рано бежать в аптеку в надежде купить новое лекарство.
Конкурс «био/мол/текст»-2018
Эта работа заняла первое место в номинации «Наглядно о ненаглядном» конкурса «био/мол/текст»-2018.
Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
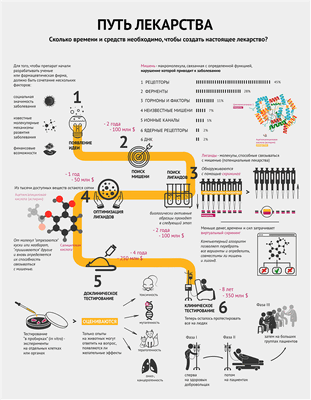
Да здравствует идея!
Для того чтобы препарат начали разрабатывать ученые или фармацевтическая фирма, должно быть сочетание нескольких факторов [1]:
- социальная значимость заболевания;
- известные молекулярные механизмы развития заболевания;
- финансовые средства и возможности по созданию конкретного лекарства.
Другими словами, должна появиться идея.
Операция «Мишень»
Совместными усилиями группа ученых выбирает мишень и способ воздействия на нее для лечения или предотвращения заболевания.
Мишень — это биологическая макромолекула, связанная с определенной функцией, нарушение которой приводит к заболеванию [2]. Чаще всего мишенями являются белки — рецепторы и ферменты. Инфографика демонстрирует, какие макромолекулы чаще всего становятся мишенями [2]. Забегая вперед, стоит отметить, что затем к мишени подбирают вещество — лекарство. Самый распространенный пример — циклооксигеназа 1 (мишень) и ацетилсалициловая кислота (аспирин) (лекарство) — тоже проиллюстрирован (см. также видео).
Видео. Лекция Валентина Табакмахера «Драг-дизайн. Современный подход к созданию лекарств».
На поиски лигандов
После того, как ученые нашли мишень, им нужно понять, чем в нее целиться. Лиганд (потенциальное лекарство) — это химическое соединение (как правило, низкомолекулярное), специфически взаимодействующее со своей мишенью и тем самым влияющее на процессы внутри клетки [2].
Исследование всех возможных веществ, конечно, нереально: существует не менее 10 40 лигандов. Поэтому на структуру потенциальных лигандов накладывают ряд ограничений, которые существенно сужают поиск. В качестве отправной точки обычно используют библиотеки соединений, которые создаются специализированными компаниями по условиям, заданным разработчиком, или уже имеются в арсенале фармацевтической фирмы. Такие библиотеки могут содержать миллионы веществ [3].
Воздействуют ли выбранные лиганды на мишень, помогает определить скрининг. Он бывает лабораторным (in vitro) или компьютерным (in silico). В случае с лабораторным скринингом на особые предметные стекла — плашки, содержащие в тысячах микролитровых лунок тестовую систему, например молекулы белка-мишени или целые клетки (при необходимости — генетически модифицированные), — робот раскапывает из пипеток исследуемые вещества, следуя заданной программе. Потом происходит считывание данных, говорящее о том, в какой лунке обнаружена биологическая активность. Детектор может определять ее по радиоактивному сигналу, флюоресценции, поляризации света и многим другим параметрам [3].
Сокращай, оптимизируй!
Из тысяч доступных веществ с более-менее определенными свойствами необходимо выбрать сотни молекул, способных после дальнейшей модификации и испытаний на бактериях или культурах клеток дать десятки так называемых кандидатных соединений, предназначенных для доклинических исследований, включая тестирование на животных.
Оптимизация может заключаться в «отсечении» части известного лиганда, или наоборот, добавлении к нему новых элементов и новой проверке на взаимодействие с мишенью. Возвращаясь к аспирину: он получился из салициловой кислоты путем добавления ацетильной группы.
Тестировали, тестировали, да вытестировали
Отобранные соединения сначала тестируются в биохимико-фармакологических исследованиях или экспериментах на клеточных культурах, изолированных клетках и изолированных органах. Так как эти модели не способны полностью воспроизвести весь комплекс биологических процессов в настоящем организме, любое потенциальное лекарство тестируется на животных. Только опыты на животных могут ответить на вопрос, появляются ли желательные эффекты в нетоксичных или малотоксичных дозах.
Исследование токсичности призвано оценить:
- токсичность при кратковременном и длительном применении;
- возможность генетических повреждений (генотоксичность, мутагенность);
- возможность развития опухолей (онко- и канцерогенность);
- возможность рождения больного плода (тератогенность).
На животных исследуемые соединения испытываются также на поглощение, распределение, метаболизм и выделение (фармакокинетика) [4].
После этого этапа отсева на стадию клинических испытаний на людях остается в лучшем случае 1−3 препарата (напомню, что изначально было примерно 1000 потенциальных лекарств!).
Выходи на рынок!
Клиническое тестирование включает в себя несколько фаз, которые иллюстрирует инфографика [5].
Сначала проводится исследование новых препаратов на здоровых лицах с целью определить, наблюдаются ли у человека эффекты, обнаруженные в тестах на животных, и выявить взаимоотношения между дозой и эффектом.
Потом потенциальный новый препарат апробируется на избранных пациентах для определения терапевтической эффективности при заболевании, для которого он предназначен. Положительное действие должно быть явным, а нежелательные эффекты приемлемо малы.
Далее к исследованию привлекаются большие группы пациентов, с помощью которых исследуемое лекарство сравнивается со стандартным лечением по исходам терапии [4].
В процессе клинических испытаний многие новые лекарства признаются негодными к применению.
Решение одобрить новый препарат принимает национальный регулирующий орган (в России — Фармкомитет МЗ РФ). Заявители (фармацевтические компании) представляют в регулирующий орган полный комплект документации преклинических и клинических испытаний, в которых полученные данные об эффективности и безопасности удовлетворяют установленным требованиям и предполагаемую форму выпуска продукта (таблетки, капсулы и т.д.)
После получения одобрения новое лекарство может продаваться под торговой маркой и, таким образом, становится доступным для назначения врачами и распространения в аптеках. Параллельно идет разработка технологического процесса производства лекарственного средства, требований к качеству, методов анализа.
По мере распространения препарата за ним продолжается наблюдение. Окончательное суждение о соотношении «польза—риск» нового лекарства может быть сделано только на основании долговременного опыта его применения. Таким образом, определяется терапевтическая ценность нового лекарственного препарата.
В разных случаях процесс разработки нового лекарства от идеи до реализации занимает примерно от 5 до 18 лет. Суммарная стоимость разработки, с учетом препаратов, не достигших рынка, часто превышает 1 млрд долларов (до 2,5 млрд в среднем) [6].
Путь лекарства — от идеи до регистрации
Как создать препарат в лаборатории и перенести его в производство
Все мы ходим в аптеку и покупаем лекарства: таблетки, порошки, мази, растворы и многие другие формы лекарственных веществ. Но мало кто знает, как происходит создание лекарств и какой путь необходимо пройти от научной разработки в руках ученого до получения регистрационного досье на готовый препарат.

Фото: Александр Коряков, Коммерсантъ / купить фото
Разработка и создание лекарственных препаратов проходят при финансовой поддержке различных государственных и коммерческих структур (фондов) в соответствии с утвержденными приоритетными направлениями развития науки, технологий и техники в Российской Федерации, согласно перечню (указ президента России от 7 июля 2011 года №899). Одним из таких направлений являются «Технологии снижения потерь от социально значимых заболеваний».
Терапия и диагностика онкологических заболеваний — одно из приоритетных направлений. Многие ученые работают над созданием новых низкомолекулярных веществ для химиотерапии, получением новых аналогов уже существующих препаратов для преодоления возникающей резистентности опухолевых клеток, а также созданием новых лекарственных форм препаратов для улучшения биодоступности активного компонента и уменьшения побочных эффектов. В последние годы широко развивается направление адресной доставки препаратов — например, на основе антител.
Нашей научной группой под руководством члена-корреспондента РАН, профессора, доктора химических наук Евгения Северина разработан универсальный подход к созданию препарата для адресной доставки в злокачественные новообразования. Данный метод заключается в синтезе наночастиц, содержащих активное вещество, с последующей конъюгацией векторной молекулой — белка, способного связываться с рецепторами на поверхности опухолевых клеток. Предварительные исследования показали многообещающие результаты, проведенные доклинические испытания подтвердили, что разработанный прототип препарата обладает большей противоопухолевой активностью по сравнению с аналогом, представленным на рынке. Суммируя опыт проведения таких исследований, мы можем описать стандартный протокол проведения исследований при разработке нового препарата и оформления нормативной документации.
Каков же общий путь исследований, позволяющих провести доказательную базу эффективности и безопасности нового препарата, и какое количество времени для этого необходимо? После проведения этапов разработки подхода создания препарата и проведения предварительных экспериментов для доказательства его эффективности коллективом оформляется заявка для участия в конкурсе для предоставления финансирования на проведение доклинических испытаний. Стандартный грант предоставляется на три года, по результатам выполнения которого у группы исследователей, выполняющих данную работу, будет готовый прототип препарата, изучена его эффективность, безопасность и оформлены все необходимые нормативные документы, на основе которых формируется досье и подается на рассмотрение в Министерство здравоохранения России.
Первое, что необходимо сделать при разработке нового препарата,— проведение обширного литературного и патентного поиска в близких и смежных областях, чтобы избежать «изобретения велосипеда». Если патентная чистота подтверждена, можно приступать к экспериментальной работе.
В полученном гранте на проведение доклинических испытаний прописан календарный план и список этапов, которые необходимо выполнить, а затем подготовить отчетную документацию. Все этапы регламентированы соответствующими нормативными документами. Настольными книгами для специалистов являются «Государственная фармакопея Российской Федерации» и «Руководство по проведению доклинических исследований лекарственных средств» под редакцией А. Н. Миронова. В фармакопее прописаны все требования и нормы к разрабатываемым препаратам, какие виды исследования необходимо провести для подтверждения состава, структуры и свойств будущего лекарства или новой лекарственной формы (порошки, таблетки, растворы и пр.). В руководстве по проведению доклинических исследований подробно изложено, как необходимо проводить доклинические испытания, чтобы исследование было стандартизировано: выбор вида животных, их количества, кратность введения, дозы и пр.
Для проведения такого широкого спектра исследований необходимо соответствующее количество будущего препарата. В лабораторных условиях обычно отрабатывают технологические режимы и оптимальные параметры получения — от температурного режима до масштабирования процесса — и изучают влияние этих параметров на свойства получаемого продукта. По оптимизированным условиям пишут лабораторный регламент, где четко описано, как именно и при каких условиях необходимо проводить каждый этап получения препарата и что должно быть на выходе, вплоть до учета потерь производства. Лабораторный регламент является официальным нормативным документом, на его основе составляют опытно-промышленный и промышленный регламенты. Для последних двух необходима специальная производственная площадка, аттестованная под изготовление похожего продукта, как и разработанный препарат (рекомбинантные белки, вакцинные препараты, противоопухолевые препараты и т. д.). Необходимо перенести лабораторную технологию получения продукта в больший масштаб на промышленное оборудование, отработать регламентированный процесс получения и оптимизировать технологию с учетом новых объемов. Таким образом, на промышленных производственных площадках по опытно-промышленному и промышленному регламенту получают опытные партии препарата, которые затем необходимо проверять на соответствие всем показателям, которые были установлены разработчиками после получения оптимальной партии по лабораторному регламенту. Все требования к полученному препарату описаны в нормативном документе — фармацевтической статье предприятия (ФСП).
ФСП пишут по тем методам, с помощью которых анализируют полученный препарат в соответствии с государственной фармакопеей. Для включения метода в ФСП необходимо использовать либо стандартизированные методы, приведенные в фармакопее, либо валидированные методы, которые были использованы, но отсутствуют в фармакопее или отличаются по условиям проведения от описанных. Чем больше активных (целевых) и вспомогательных компонентов в препарате, тем больше методов содержит ФСП. Необходимо провести количественный анализ активного компонента и примесей, полный качественный анализ, а также подтвердить сохранение функциональной активности основного компонента.
Зачем нужна фармацевтическая статья предприятия? В технологическом процессе получения препарата могут возникнуть непредвиденные неполадки на какой-либо стадии производства. Для выявления несоответствия продукта (брака производства) необходимо проводить анализ каждой партии. Если была получена бракованная партия, ее можно будет легко выявить, проведя все анализы и сравнив с установленными нормами в ФСП.
После получения опытных партий необходимо изучить стабильность полученного препарата, чтобы доказать, что за указанный промежуток времени (срок годности) не происходит никаких существенных изменений и свойства будущего лекарства остаются неизменными. Стандартный срок хранения лекарственных средств — от полугода до трех лет. Для противоопухолевых препаратов — два года. Для конкурентоспособности и рентабельности будущего лекарства необходимо придерживаться таких же сроков годности, а также ориентироваться на аналоги. Однако ждать два года, чтобы узнать, стабилен ли препарат и проходит ли он по всем нормам и стандартам, нет необходимости. Существуют протоколы, описанные в фармакопее (ОФС.1.1.0009.15 «Сроки годности лекарственных средств»), позволяющие сократить период исследования до года и даже шести месяцев, используя более агрессивные условия исследования. Если препарат по всем показателям сохраняет количественные, качественные и функциональные характеристики, указанные в ФСП, следующий этап — проведение доклинических исследований на животных. На все перечисленные стадии оформляют нормативные документы: регламенты на производство, отчеты о валидации, акты наработки, ФСП, протоколы анализа партий, подтверждающие соответствие полученного продукта описанному в документах.
Для изучения безопасности и эффективности полученного препарата на лабораторных животных утверждается план доклинических исследований, в котором перечислены все этапы исследования и их последовательность. План и модели проведения исследований составляются в зависимости от типа разработанного препарата и описаны в «Руководстве по проведению доклинических исследований лекарственных средств». Для оформления регистрационного досье необходимо провести полные доклинические исследования: исследование нескольких видов токсичности (общетоксическое действие, аллергизирующие свойства, иммунотоксическое действие, репродуктивная токсичность и др.), эффективности действия (например, противоопухолевый эффект нового препарата в сравнении с аналогами), изучить фармакокинетику. Все полученные данные обрабатывают статистически и оформляют в нормативный документ — отчет о доклинических исследованиях с прикреплением первичных результатов. На основании полученных данных, в случае если препарат обладает эффективностью и при этом безопасен для применения, разработчики пишут план проведения первой стадии клинических испытаний, проект инструкции по применению и проект брошюры исследователя. Из составленных документов формируется регистрационное досье, которое и подается на рассмотрение в Министерство здравоохранения России с другими сопутствующими документами.
До последнего этапа, описанного в данной статье, доходят немногие разработки. Путь от научной идеи до регистрации может занимать от трех лет до десятилетий. При наличии оформившейся идеи, прошедшей предварительные фундаментальные исследования, все описанные этапы исследования можно провести за три-пять лет. А дальше препарат ждет еще более сложный, но не менее интересный путь: клинические испытания, регистрация, производство и выход на рынок — при условии наличия хороших результатов на этапе клинических испытаний.
Елена Никольская, кандидат химических наук, старший научный сотрудник ИБХФ РАН
Читайте также:
- Объем мертвого пространства. Мертвое пространство дыхательного аппарата
- Хронический отек тыла кисти после травмы
- Шейный спондилез: причины, симптомы и лечение
- Непереживаемые эмоции. Основа или морфология эмоций
- Доступ и ход операции дивертикуляризации двенадцатиперстной кишки, панкреатического дренажа при травме